ERBB2 Influences the Subcellular Localization of the Estrogen
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supplementary Information Material and Methods
MCT-11-0474 BKM120: a potent and specific pan-PI3K inhibitor Supplementary Information Material and methods Chemicals The EGFR inhibitor NVP-AEE788 (Novartis), the Jak inhibitor I (Merck Calbiochem, #420099) and anisomycin (Alomone labs, # A-520) were prepared as 50 mM stock solutions in 100% DMSO. Doxorubicin (Adriablastin, Pfizer), EGF (Sigma Ref: E9644), PDGF (Sigma, Ref: P4306) and IL-4 (Sigma, Ref: I-4269) stock solutions were prepared as recommended by the manufacturer. For in vivo administration: Temodal (20 mg Temozolomide capsules, Essex Chemie AG, Luzern) was dissolved in 4 mL KZI/glucose (20/80, vol/vol); Taxotere was bought as 40 mg/mL solution (Sanofi Aventis, France), and prepared in KZI/glucose. Antibodies The primary antibodies used were as follows: anti-S473P-Akt (#9271), anti-T308P-Akt (#9276,), anti-S9P-GSK3β (#9336), anti-T389P-p70S6K (#9205), anti-YP/TP-Erk1/2 (#9101), anti-YP/TP-p38 (#9215), anti-YP/TP-JNK1/2 (#9101), anti-Y751P-PDGFR (#3161), anti- p21Cip1/Waf1 (#2946), anti-p27Kip1 (#2552) and anti-Ser15-p53 (#9284) antibodies were from Cell Signaling Technologies; anti-Akt (#05-591), anti-T32P-FKHRL1 (#06-952) and anti- PDGFR (#06-495) antibodies were from Upstate; anti-IGF-1R (#SC-713) and anti-EGFR (#SC-03) antibodies were from Santa Cruz; anti-GSK3α/β (#44610), anti-Y641P-Stat6 (#611566), anti-S1981P-ATM (#200-301), anti-T2609 DNA-PKcs (#GTX24194) and anti- 1 MCT-11-0474 BKM120: a potent and specific pan-PI3K inhibitor Y1316P-IGF-1R were from Bio-Source International, Becton-Dickinson, Rockland, GenTex and internal production, respectively. The 4G10 antibody was from Millipore (#05-321MG). -
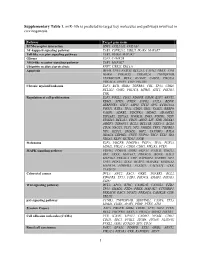
1 Supplementary Table 1. Mir-10B Is Predicted to Target Key Molecules
Supplementary Table 1. miR-10b is predicted to target key molecules and pathways involved in carcinogenesis. Pathway Target gene name ECM-receptor interaction SDC1, COL24A1, COL4A4 NF-kappa B signaling pathway TAB1, CSNK2A2, UBE2I, IRAK4, MAP3K7 Toll-like receptor signaling pathway TAB1, IRAK4, MAP3K7 Glioma E2F3, CAMK2B NOD-like receptor signaling pathway TAB1, MAP3K7 Ubiquitin mediated proteolysis RNF7, UBE2I, ERCC8 Apoptosis DFFB, TP53, FASLG, BCL2L1, CAPN2, PRKX, ATM, IRAK4, PRKACG, PRKAR2A, TNFRSF10B, TNFRSF10D, BCL2, IL1RAP, CASP8, PIK3CA, PRKACA, APAF1, CHP, PIK3R3 Chronic myeloid leukemia E2F3, BCR, GRB2, TGFBR1, CBL, TP53, CDK6, BCL2L1, GAB2, PIK3CA, MDM2, SHC1, PIK3R3, CRK Regulation of cell proliferation E2F3, FOSL2, CDX2, PDGFB, OSMR, E2F7, ARNT2, RBM5, STRN, PTEN, S1PR2, CUL3, BDNF, SERPINE1, SHC1, ASPH, ITCH, SPN, CCDC88A, FOXJ1, RXRA, TP53, CDK6, IRS1, VASH2, RBBP9, VASH1, ADRB2, PDGFRA, MDM2, ADAMTS1, EIF2AK2, EIF5A2, ICOSLG, ING5, FGFR3, NDN, ST8SIA1, BCL2L1, CDH5, ARNT, LIF, VDR, HOXA3, AGGF1, TSPAN31, BCL2, BCL11B, NKX3-1, BCL6, CD28, NACC1, FLT1, NF2, JARID2, TBX5, TGFBR1, NF1, KLF11, SMAD2, IGF2, TAX1BP3, BTLA, HDAC4, LEPRE1, CNTF, NUP62, TSC1, ETS1, ID4, NR5A2, KLF4, KCTD11, NFIB Melanoma E2F3, PDGFB, PDGFRA, FGF11, TP53, FGF23, MDM2, PIK3CA, CDK6, CDH1, PIK3R3, PTEN MAPK signaling pathway FGFR3, PDGFB, GRB2, FGF11, FASLG, GNG12, SRF, PRKX, MAP3K7, PRKACG, BDNF, RAC3, MAP3K2, PRKACA, CHP, RAPGEF2, TGFBR1, NF1, TP53, FGF23, STK4, DUSP5, MAP4K4, RPS6KA2, MAPK14, PDGFRA, PLA2G3, CACNA1C, CRK, PLA2G2F Colorectal cancer -

The P90 RSK Family Members: Common Functions and Isoform Specificity
Published OnlineFirst August 22, 2013; DOI: 10.1158/0008-5472.CAN-12-4448 Cancer Review Research The p90 RSK Family Members: Common Functions and Isoform Specificity Romain Lara, Michael J. Seckl, and Olivier E. Pardo Abstract The p90 ribosomal S6 kinases (RSK) are implicated in various cellular processes, including cell proliferation, survival, migration, and invasion. In cancer, RSKs modulate cell transformation, tumorigenesis, and metastasis. Indeed, changes in the expression of RSK isoforms have been reported in several malignancies, including breast, prostate, and lung cancers. Four RSK isoforms have been identified in humans on the basis of their high degree of sequence homology. Although this similarity suggests some functional redundancy between these proteins, an increasing body of evidence supports the existence of isoform-based specificity among RSKs in mediating particular cellular processes. This review briefly presents the similarities between RSK family members before focusing on the specific function of each of the isoforms and their involvement in cancer progression. Cancer Res; 73(17); 1–8. Ó2013 AACR. Introduction subsequently cloned throughout the Metazoan kingdom (2). The extracellular signal–regulated kinase (ERK)1/2 pathway The genomic analysis of several cancer types suggests that fi is involved in key cellular processes, including cell prolifera- these genes are not frequently ampli ed or mutated, with some tion, differentiation, survival, metabolism, and migration. notable exceptions (e.g., in the case of hepatocellular carcino- More than 30% of all human cancers harbor mutations within ma; ref. 6). Table 1 summarizes reported genetic changes in this pathway, mostly resulting in gain of function and conse- RSK genes. -

CPTC-MAPK3-1 (CAB079934) Immunohistochemistry
CPTC-MAPK3-1 (CAB079934) Uniprot ID: P27361 Protein name: MK03_HUMAN Full name: Mitogen-activated protein kinase 3 Function: Serine/threonine kinase which acts as an essential component of the MAP kinase signal transduction pathway. MAPK1/ERK2 and MAPK3/ERK1 are the 2 MAPKs which play an important role in the MAPK/ERK cascade. They participate also in a signaling cascade initiated by activated KIT and KITLG/SCF. Depending on the cellular context, the MAPK/ERK cascade mediates diverse biological functions such as cell growth, adhesion, survival and differentiation through the regulation of transcription, translation, cytoskeletal rearrangements. The MAPK/ERK cascade plays also a role in initiation and regulation of meiosis, mitosis, and postmitotic functions in differentiated cells by phosphorylating a number of transcription factors. About 160 substrates have already been discovered for ERKs. Many of these substrates are localized in the nucleus, and seem to participate in the regulation of transcription upon stimulation. However, other substrates are found in the cytosol as well as in other cellular organelles, and those are responsible for processes such as translation, mitosis and apoptosis. Moreover, the MAPK/ERK cascade is also involved in the regulation of the endosomal dynamics, including lysosome processing and endosome cycling through the perinuclear recycling compartment (PNRC); as well as in the fragmentation of the Golgi apparatus during mitosis. The substrates include transcription factors (such as ATF2, BCL6, ELK1, ERF, FOS, HSF4 or SPZ1), cytoskeletal elements (such as CANX, CTTN, GJA1, MAP2, MAPT, PXN, SORBS3 or STMN1), regulators of apoptosis (such as BAD, BTG2, CASP9, DAPK1, IER3, MCL1 or PPARG), regulators of translation (such as EIF4EBP1) and a variety of other signaling-related molecules (like ARHGEF2, FRS2 or GRB10). -

Comparison of the Differential Expression Mirnas in Wistar Rats Before and 10 Days After S. Japonicum Infection
Han et al. Parasites & Vectors 2013, 6:120 http://www.parasitesandvectors.com/content/6/1/120 RESEARCH Open Access Comparison of the differential expression miRNAs in Wistar rats before and 10 days after S.japonicum infection Hongxiao Han1,2†, Jinbiao Peng1,3†, Yang Hong1, Min Zhang1, Yanhui Han1, Zhiqiang Fu1, Yaojun Shi1, Jinjun Xu2, Jianping Tao2* and Jiaojiao Lin1* Abstract Background: When compared to the murine permissive host of Schistosoma japonicum, Wistar rats are less susceptible to Schistosoma japonicum infection, and are considered to provide a less suitable microenvironment for parasite growth and development. MicroRNAs (miRNAs), are a class of endogenous, non-coding small RNAs, that impose an additional, highly significant, level of gene regulation within eukaryotes. Methods: To investigate the regulatory mechanisms provided by miRNA in the schistosome-infected rat model, we utilized a miRNA microarray to compare the progression of miRNA expression within different host tissues both before and 10 days after cercarial infection, in order to identify potential miRNAs with roles in responding to a schistosome infection. Results: Among the analysed miRNAs, 16 within the liver, 61 within the spleen and 10 within the lung, were differentially expressed in infected Wistar rats. Further analysis of the differentially expressed miRNAs revealed that many important signal pathways are triggered after infection with S. japonicum in Wistar rats. These include the signal transduction mechanisms associated with the Wnt and MAPK signaling pathways, cellular differentiation, with a particular emphasis on adipocyte and erythroid differentiation. Conclusions: The results presented here include the identification of specific differentially expressed miRNAs within the liver, lungs and spleen of Wistar rats. -

PRODUCTS and SERVICES Target List
PRODUCTS AND SERVICES Target list Kinase Products P.1-11 Kinase Products Biochemical Assays P.12 "QuickScout Screening Assist™ Kits" Kinase Protein Assay Kits P.13 "QuickScout Custom Profiling & Panel Profiling Series" Targets P.14 "QuickScout Custom Profiling Series" Preincubation Targets Cell-Based Assays P.15 NanoBRET™ TE Intracellular Kinase Cell-Based Assay Service Targets P.16 Tyrosine Kinase Ba/F3 Cell-Based Assay Service Targets P.17 Kinase HEK293 Cell-Based Assay Service ~ClariCELL™ ~ Targets P.18 Detection of Protein-Protein Interactions ~ProbeX™~ Stable Cell Lines Crystallization Services P.19 FastLane™ Structures ~Premium~ P.20-21 FastLane™ Structures ~Standard~ Kinase Products For details of products, please see "PRODUCTS AND SERVICES" on page 1~3. Tyrosine Kinases Note: Please contact us for availability or further information. Information may be changed without notice. Expression Protein Kinase Tag Carna Product Name Catalog No. Construct Sequence Accession Number Tag Location System HIS ABL(ABL1) 08-001 Full-length 2-1130 NP_005148.2 N-terminal His Insect (sf21) ABL(ABL1) BTN BTN-ABL(ABL1) 08-401-20N Full-length 2-1130 NP_005148.2 N-terminal DYKDDDDK Insect (sf21) ABL(ABL1) [E255K] HIS ABL(ABL1)[E255K] 08-094 Full-length 2-1130 NP_005148.2 N-terminal His Insect (sf21) HIS ABL(ABL1)[T315I] 08-093 Full-length 2-1130 NP_005148.2 N-terminal His Insect (sf21) ABL(ABL1) [T315I] BTN BTN-ABL(ABL1)[T315I] 08-493-20N Full-length 2-1130 NP_005148.2 N-terminal DYKDDDDK Insect (sf21) ACK(TNK2) GST ACK(TNK2) 08-196 Catalytic domain -

The Serine/Threonine Protein Kinase (Akt)/ Protein Kinase B (Pkb) Signaling Pathway in Breast Cancer
Journal of Mind and Medical Sciences Volume 7 Issue 1 Article 7 2020 The Serine/Threonine Protein Kinase (Akt)/ Protein Kinase B (PkB) Signaling Pathway in Breast Cancer Daniela Miricescu FACULTY OF DENTAL MEDICINE, DEPARTMENT OF BIOCHEMISTRY, BUCHAREST, ROMANIA Camelia Cristina Diaconu CAROL DAVILA UNIVERSITY OF MEDICINE AND PHARMACY, CLINICAL EMERGENCY HOSPITAL OF BUCHAREST, INTERNAL MEDICINE CLINIC, BUCHAREST, ROMANIA Constantin Stefani CAROL DAVILA UNIVERSITY OF MEDICINE AND PHARMACY, DEPARTMENT OF FAMILY MEDICINE, BUCHAREST, ROMANIA Ana Maria Alexandra Stanescu CAROL DAVILA UNIVERSITY OF MEDICINE AND PHARMACY, DEPARTMENT OF FAMILY MEDICINE, BUCHAREST, ROMANIA Alexandra Totan FAollowCULT thisY OF and DEN additionalTAL MEDICINE, works at:DEP https:/ARTMEN/scholarT OF.v BIOCHEMISTRalpo.edu/jmmsY, BUCHAREST, ROMANIA Part of the Obstetrics and Gynecology Commons, Oncology Commons, and the Palliative Care SeeCommons next page for additional authors Recommended Citation Miricescu, Daniela; Diaconu, Camelia Cristina; Stefani, Constantin; Stanescu, Ana Maria Alexandra; Totan, Alexandra; Rusu, Ioana Ruxandra; Bratu, Ovidiu Gabriel; Spinu, Dan; and Greabu, Maria (2020) "The Serine/ Threonine Protein Kinase (Akt)/ Protein Kinase B (PkB) Signaling Pathway in Breast Cancer," Journal of Mind and Medical Sciences: Vol. 7 : Iss. 1 , Article 7. DOI: 10.22543/7674.71.P3439 Available at: https://scholar.valpo.edu/jmms/vol7/iss1/7 This Review Article is brought to you for free and open access by ValpoScholar. It has been accepted for inclusion in Journal -
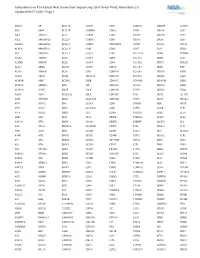
Comprehensive Pan-Cancer Next Generation Sequencing Solid Tumor Panel, Aberration List Updated 08-07-2020 --Page 1
Comprehensive Pan-Cancer Next Generation Sequencing Solid Tumor Panel, Aberration List Updated 08-07-2020 --Page 1 ABCC3 AR BCL11A CANT1 CDK1 CMKLR1 DAB2IP DUSP9 ABI1 ARAF BCL11B CAPRIN1 CDK12 CNBP DACH1 E2F1 ABL1 ARFRP1 BCL2 CAPZB CDK2 CNOT2 DACH2 E2F3 ABL2 ARHGAP20 BCL2A1 CARD11 CDK4 CNTN1 DAXX EBF1 ABLIM1 ARHGAP26 BCL2L1 CARM1 CDK5RAP2 CNTRL DCLK2 ECT2L ACACA ARHGEF12 BCL2L11 CARS CDK6 COG5 DCN EDIL3 ACE ARHGEF7 BCL2L2 CASC5 CDK7 COL11A1 DDB1 EDNRB ACER1 ARID1A BCL3 CASP3 CDK8 COL1A1 DDB2 EED ACSBG1 ARID1B BCL6 CASP7 CDK9 COL1A2 DDIT3 EEFSEC ACSL3 ARID2 BCL7A CASP8 CDKL5 COL3A1 DDR2 EGF ACSL6 ARID5B BCL9 CAV1 CDKN1A COL6A3 DDX10 EGFR ACVR1 ARIH2 BCOR CBFA2T3 CDKN1B COL9A3 DDX20 EGR1 ACVR1B ARNT BCORL1 CBFB CDKN1C COMMD1 DDX39B EGR2 ACVR1C ARRDC4 BCR CBL CDKN2A COX6C DDX3X EGR3 ACVR2A ASMTL BDNF CBLB CDKN2B CPNE1 DDX41 EGR4 ADD3 ASPH BHLHE22 CBLC CDKN2C CPS1 DDX5 EIF1AX ADM ASPSCR1 BICC1 CCDC28A CDKN2D CPSF6 DDX6 EIF4A2 AFF1 ASTN2 BIN1 CCDC6 CDX1 CRADD DEK EIF4E AFF3 ASXL1 BIRC3 CCDC88C CDX2 CREB1 DGKB ELF3 AFF4 ASXL2 BIRC6 CCK CEBPA CREB3L1 DGKI ELF4 AGR3 ATF1 BLM CCL2 CEBPB CREB3L2 DGKZ ELK4 AHCYL1 ATF3 BMP4 CCNA2 CEBPD CREBBP DICER1 ELL AHI1 ATG13 BMPR1A CCNB1IP1 CEBPE CRKL DIRAS3 ELN AHR ATG5 BRAF CCNB3 CENPF CRLF2 DIS3 ELOVL2 AHRR ATIC BRCA1 CCND1 CENPU CRTC1 DIS3L2 ELP2 AIP ATL1 BRCA2 CCND2 CEP170B CRTC3 DKK1 EML1 AK2 ATM BRCC3 CCND3 CEP57 CSF1 DKK2 EML4 AK5 ATP1B4 BRD1 CCNE1 CEP85L CSF1R DKK4 ENPP2 AKAP12 ATP8A2 BRD3 CCNG1 CHCHD7 CSF3 DLEC1 EP300 AKAP6 ATR BRD4 CCT6B CHD2 CSF3R DLL1 EP400 AKAP9 ATRNL1 BRIP1 CD19 CHD4 CSNK1A1 DLL3 -

Targeting the Hsp90-Cdc37-Client Protein Interaction to Disrupt Hsp90 Chaperone Machinery Ting Li1, Hu-Lin Jiang2, Yun-Guang Tong3,4 and Jin-Jian Lu1*
Li et al. Journal of Hematology & Oncology (2018) 11:59 https://doi.org/10.1186/s13045-018-0602-8 REVIEW Open Access Targeting the Hsp90-Cdc37-client protein interaction to disrupt Hsp90 chaperone machinery Ting Li1, Hu-Lin Jiang2, Yun-Guang Tong3,4 and Jin-Jian Lu1* Abstract Heat shock protein 90 (Hsp90) is a critical molecular chaperone protein that regulates the folding, maturation, and stability of a wide variety of proteins. In recent years, the development of Hsp90-directed inhibitors has grown rapidly, and many of these inhibitors have entered clinical trials. In parallel, the functional dissection of the Hsp90 chaperone machinery has highlighted the activity disruption of Hsp90 co-chaperone as a potential target. With the roles of Hsp90 co-chaperones being elucidated, cell division cycle 37 (Cdc37), a ubiquitous co-chaperone of Hsp90 that directs the selective client proteins into the Hsp90 chaperone cycle, shows great promise. Moreover, the Hsp90-Cdc37-client interaction contributes to the regulation of cellular response and cellular growth and is more essential to tumor tissues than normal tissues. Herein, we discuss the current understanding of the clients of Hsp90-Cdc37, the interaction of Hsp90-Cdc37-client protein, and the therapeutic possibilities of targeting Hsp90-Cdc37-client protein interaction as a strategy to inhibit Hsp90 chaperone machinery to present new insights on alternative ways of inhibiting Hsp90 chaperone machinery. Keywords: Hsp90 chaperone machinery, Cdc37, Kinase client, Protein interaction Background chloroplast HSP90C, mitochondrial TNFR-associated protein, Heat shock protein 90 (Hsp90) is a critically conserved and bacterial high-temperature protein G [2, 8]. In this protein and one of the major molecular chaperones within review,weusethetermHsp90torefertotheseHsp90 eukaryotic cells [1]. -

MAPK3 Antibody (C-Term) Blocking Peptide Synthetic Peptide Catalog # Bp7251b
10320 Camino Santa Fe, Suite G San Diego, CA 92121 Tel: 858.875.1900 Fax: 858.622.0609 MAPK3 Antibody (C-term) Blocking Peptide Synthetic peptide Catalog # BP7251b Specification MAPK3 Antibody (C-term) Blocking MAPK3 Antibody (C-term) Blocking Peptide - Peptide - Background Product Information MAPK3 is a member of the MAP kinase family. Primary Accession P27361 MAP kinases, also known as extracellular signal-regulated kinases (ERKs), act in a signaling cascade thatregulates various MAPK3 Antibody (C-term) Blocking Peptide - Additional Information cellular processes such as proliferation, differentiation, and cell cycle progression in response to a variety of extracellular signals. Gene ID 5595 This kinase is activated by upstream kinases, resulting in its translocation to the nucleus Other Names where it phosphorylates nuclear targets. Mitogen-activated protein kinase 3, MAP kinase 3, MAPK 3, ERT2, Extracellular MAPK3 Antibody (C-term) Blocking signal-regulated kinase 1, ERK-1, Peptide - References Insulin-stimulated MAP2 kinase, MAP kinase isoform p44, p44-MAPK, Microtubule-associated protein 2 kinase, Munshi, H.G., et al., J. Biol. Chem. p44-ERK1, MAPK3, ERK1, PRKM3 279(37):39042-39050 (2004).Mukherjee, S., et al., Infect. Immun. 72(9):5274-5282 Target/Specificity (2004).Sebkova, L., et al., Infect. Immun. The synthetic peptide sequence used to 72(9):5019-5026 (2004).Huang, H.M., et al., generate the antibody <a href=/product/pr Biochem. Biophys. Res. Commun. oducts/AP7251b>AP7251b</a> was 320(4):1247-1252 (2004).Mizuno, S., et al., selected from the C-term region of human Am. J. Respir. Cell Mol. Biol. 31(2):184-192 MAPK3. -

SUPPLEMENTAL MATERIAL Acknowledgments
SUPPLEMENTAL MATERIAL Acknowledgments The members of the CARDIoGRAM consortium are: Heribert Schunkert, Inke R. König, Sekar Kathiresan, Muredach P. Reilly, Themistocles L. Assimes, Hilma Holm, Michael Preuss, Alexandre F. R. Stewart, Maja Barbalic, Christian Gieger, Devin Absher, Zouhair Aherrahrou, Hooman Allayee, David Altshuler, Sonia S. Anand, Karl Andersen, Jeffrey L. Anderson, Diego Ardissino, Stephen G. Ball, Anthony J. Balmforth, Timothy A. Barnes, Diane M. Becker, Lewis C. Becker, Klaus Berger, Joshua C. Bis, S. Matthijs Boekholdt, Eric Boerwinkle, Peter S. Braund, Morris J. Brown, Mary Susan Burnett, Ian Buysschaert, Cardiogenics, John F. Carlquist, Li Chen, Sven Cichon, Veryan Codd, Robert W. Davies, George Dedoussis, Abbas Dehghan, Serkalem Demissie, Joseph M. Devaney, Ron Do, Angela Doering, Sandra Eifert, Nour Eddine El Mokhtari, Stephen G. Ellis, Roberto Elosua, James C. Engert, Stephen E. Epstein, Ulf de Faire, Marcus Fischer, Aaron R. Folsom, Jennifer Freyer, Bruna Gigante, Domenico Girelli, Solveig Gretarsdottir, Vilmundur Gudnason, Jeffrey R. Gulcher, Eran Halperin, Naomi Hammond, Stanley L. Hazen, Albert Hofman, Benjamin D. Horne, Thomas Illig, Carlos Iribarren, Gregory T. Jones, J.Wouter Jukema, Michael A. Kaiser, Lee M. Kaplan, John J.P. Kastelein, Kay-Tee Khaw, Joshua W. Knowles, Genovefa Kolovou, Augustine Kong, Reijo Laaksonen, Diether Lambrechts, Karin Leander, Guillaume Lettre, Mingyao Li, Wolfgang Lieb, Patrick Linsel-Nitschke, Christina Loley, Andrew J. Lotery, Pier M. Mannucci, Seraya Maouche, Nicola Martinelli, Pascal P. McKeown, Christa Meisinger, Thomas Meitinger, Olle Melander, Pier Angelica Merlini, Vincent Mooser, Thomas Morgan, Thomas W. Mühleisen, Joseph B. Muhlestein, Thomas Münzel, Kiran Musunuru, Janja Nahrstaedt, Christopher P. Nelson, Markus M. Nöthen, Oliviero Olivieri, Riyaz S. -

RPS6KA2 Rabbit Pab
Leader in Biomolecular Solutions for Life Science RPS6KA2 Rabbit pAb Catalog No.: A16305 Basic Information Background Catalog No. This gene encodes a member of the RSK (ribosomal S6 kinase) family of A16305 serine/threonine kinases. This kinase contains two non-identical kinase catalytic domains and phosphorylates various substrates, including members of the mitogen- Observed MW activated kinase (MAPK) signalling pathway. The activity of this protein has been 83kDa implicated in controlling cell growth and differentiation. Alternative splice variants, encoding different isoforms, have been characterized. Calculated MW 83kDa Category Primary antibody Applications WB,IHC,IF Cross-Reactivity Human, Mouse, Rat Recommended Dilutions Immunogen Information WB 1:500 - 1:2000 Gene ID Swiss Prot 6196 Q15349 IHC 1:100 - 1:200 Immunogen 1:50 - 1:200 IF Recombinant fusion protein containing a sequence corresponding to amino acids 632-733 of human RPS6KA2 (NP_066958.2). Synonyms RPS6KA2;HU-2;MAPKAPK1C;RSK;RSK3;S6K-alpha;S6K-alpha2;p90- RSK3;p90RSK2;pp90RSK3 Contact Product Information www.abclonal.com Source Isotype Purification Rabbit IgG Affinity purification Storage Store at -20℃. Avoid freeze / thaw cycles. Buffer: PBS with 0.02% sodium azide,50% glycerol,pH7.3. Validation Data Western blot analysis of extracts of various cell lines, using RPS6KA2 antibody (A16305) at 1:1000 dilution. Secondary antibody: HRP Goat Anti-Rabbit IgG (H+L) (AS014) at 1:10000 dilution. Lysates/proteins: 25ug per lane. Blocking buffer: 3% nonfat dry milk in TBST. Detection: ECL Basic Kit (RM00020). Exposure time: 30s. Immunohistochemistry of paraffin- Immunohistochemistry of paraffin- Immunofluorescence analysis of U-2 OS embedded human placenta using embedded mouse spleen using RPS6KA2 cells using RPS6KA2 antibody (A16305) at RPS6KA2 antibody (A16305) at dilution of antibody (A16305) at dilution of 1:100 (40x dilution of 1:100.