SUPPLEMENTAL MATERIAL Acknowledgments
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

DNA Methylation of GHSR, GNG4, HOXD9 and SALL3 Is a Common Epigenetic Alteration in Thymic Carcinoma
INTERNATIONAL JOURNAL OF ONCOLOGY 56: 315-326, 2020 DNA methylation of GHSR, GNG4, HOXD9 and SALL3 is a common epigenetic alteration in thymic carcinoma REINA KISHIBUCHI1, KAZUYA KONDO1, SHIHO SOEJIMA1, MITSUHIRO TSUBOI2, KOICHIRO KAJIURA2, YUKIKIYO KAWAKAMI2, NAOYA KAWAKITA2, TORU SAWADA2, HIROAKI TOBA2, MITSUTERU YOSHIDA2, HIROMITSU TAKIZAWA2 and AKIRA TANGOKU2 1Department of Oncological Medical Services, Graduate School of Biomedical Sciences, Tokushima University, Tokushima 770-8509; 2Department of Thoracic, Endocrine Surgery and Oncology, Graduate School of Biomedical Sciences, Tokushima University, Tokushima 770-8503, Japan Received August 17, 2019; Accepted October 25, 2019 DOI: 10.3892/ijo.2019.4915 Abstract. Thymic epithelial tumors comprise thymoma, promoter methylation of the 4 genes was not significantly thymic carcinoma and neuroendocrine tumors of the thymus. higher in advanced-stage tumors than in early-stage tumors in Recent studies have revealed that the incidence of somatic all thymic epithelial tumors. Among the 4 genes, relapse-free non‑synonymous mutations is significantly higher in thymic survival was significantly worse in tumors with a higher DNA carcinoma than in thymoma. However, limited information methylation than in those with a lower DNA methylation in all is currently available on epigenetic alterations in these types thymic epithelial tumors. Moreover, relapse-free survival was of cancer. In this study, we thus performed genome-wide significantly worse in thymomas with a higher DNA methyla- screening of aberrantly methylated CpG islands in thymoma tion of HOXD9 and SALL3 than in those with a lower DNA and thymic carcinoma using Illumina HumanMethylation450 methylation. On the whole, the findings of this study indicated K BeadChip. We identified 92 CpG islands significantly that the promoter methylation of cancer-related genes was hypermethylated in thymic carcinoma in relation to thymoma significantly higher in thymic carcinoma than in thymoma and and selected G protein subunit gamma 4 (GNG4), growth the thymus. -

Mechanisms Governing Anaphylaxis: Inflammatory Cells, Mediators
International Journal of Molecular Sciences Review Mechanisms Governing Anaphylaxis: Inflammatory Cells, Mediators, Endothelial Gap Junctions and Beyond Samantha Minh Thy Nguyen 1, Chase Preston Rupprecht 2, Aaisha Haque 3, Debendra Pattanaik 4, Joseph Yusin 5 and Guha Krishnaswamy 1,3,* 1 Department of Medicine, Wake Forest School of Medicine, Winston-Salem, NC 27106, USA; [email protected] 2 The Rowan School of Osteopathic Medicine, Stratford, NJ 08084, USA; [email protected] 3 The Bill Hefner VA Medical Center, Salisbury, NC 27106, USA; [email protected] 4 Division of Allergy and Immunology, UT Memphis College of Medicine, Memphis, TN 38103, USA; [email protected] 5 The Division of Allergy and Immunology, Greater Los Angeles VA Medical Center, Los Angeles, CA 90011, USA; [email protected] * Correspondence: [email protected] Abstract: Anaphylaxis is a severe, acute, life-threatening multisystem allergic reaction resulting from the release of a plethora of mediators from mast cells culminating in serious respiratory, cardiovascular and mucocutaneous manifestations that can be fatal. Medications, foods, latex, exercise, hormones (progesterone), and clonal mast cell disorders may be responsible. More recently, novel syndromes such as delayed reactions to red meat and hereditary alpha tryptasemia have been described. Anaphylaxis manifests as sudden onset urticaria, pruritus, flushing, erythema, Citation: Nguyen, S.M.T.; Rupprecht, angioedema (lips, tongue, airways, periphery), myocardial dysfunction (hypovolemia, distributive -

Product Data Sheet
Product Data Sheet ExProfileTM Human AMPK Signaling Related Gene qPCR Array For focused group profiling of human AMPK signaling genes expression Cat. No. QG004-A (4 x 96-well plate, Format A) Cat. No. QG004-B (4 x 96-well plate, Format B) Cat. No. QG004-C (4 x 96-well plate, Format C) Cat. No. QG004-D (4 x 96-well plate, Format D) Cat. No. QG004-E (4 x 96-well plate, Format E) Plates available individually or as a set of 6. Each set contains 336 unique gene primer pairs deposited in one 96-well plate. Introduction The ExProfile human AMPK signaling related gene qPCR array profiles the expression of 336 human genes related to AMPK-mediated signal transduction. These genes are carefully chosen for their close pathway correlation based on a thorough literature search of peer-reviewed publications, mainly including genes that encode AMP-activated protein kinase complex,its regulators and targets involved in many important biological processes, such as glucose uptake, β-oxidation of fatty acids and modulation of insulin secretion. This array allows researchers to study the pathway-related genes to gain understanding of their roles in the different biological processes. QG004 plate 01: 84 unique gene PCR primer pairs QG004 plate 02: 84 unique gene PCR primer pairs QG004 plate 03: 84 unique gene PCR primer pairs QG004 plate 04: 84 unique gene PCR primer pairs Shipping and storage condition Shipped at room temperate Stable for at least 6 months when stored at -20°C Array format GeneCopoeia provides five qPCR array formats (A, B, C, D, and E) suitable for use with the following real- time cyclers. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Supplementary Information Material and Methods
MCT-11-0474 BKM120: a potent and specific pan-PI3K inhibitor Supplementary Information Material and methods Chemicals The EGFR inhibitor NVP-AEE788 (Novartis), the Jak inhibitor I (Merck Calbiochem, #420099) and anisomycin (Alomone labs, # A-520) were prepared as 50 mM stock solutions in 100% DMSO. Doxorubicin (Adriablastin, Pfizer), EGF (Sigma Ref: E9644), PDGF (Sigma, Ref: P4306) and IL-4 (Sigma, Ref: I-4269) stock solutions were prepared as recommended by the manufacturer. For in vivo administration: Temodal (20 mg Temozolomide capsules, Essex Chemie AG, Luzern) was dissolved in 4 mL KZI/glucose (20/80, vol/vol); Taxotere was bought as 40 mg/mL solution (Sanofi Aventis, France), and prepared in KZI/glucose. Antibodies The primary antibodies used were as follows: anti-S473P-Akt (#9271), anti-T308P-Akt (#9276,), anti-S9P-GSK3β (#9336), anti-T389P-p70S6K (#9205), anti-YP/TP-Erk1/2 (#9101), anti-YP/TP-p38 (#9215), anti-YP/TP-JNK1/2 (#9101), anti-Y751P-PDGFR (#3161), anti- p21Cip1/Waf1 (#2946), anti-p27Kip1 (#2552) and anti-Ser15-p53 (#9284) antibodies were from Cell Signaling Technologies; anti-Akt (#05-591), anti-T32P-FKHRL1 (#06-952) and anti- PDGFR (#06-495) antibodies were from Upstate; anti-IGF-1R (#SC-713) and anti-EGFR (#SC-03) antibodies were from Santa Cruz; anti-GSK3α/β (#44610), anti-Y641P-Stat6 (#611566), anti-S1981P-ATM (#200-301), anti-T2609 DNA-PKcs (#GTX24194) and anti- 1 MCT-11-0474 BKM120: a potent and specific pan-PI3K inhibitor Y1316P-IGF-1R were from Bio-Source International, Becton-Dickinson, Rockland, GenTex and internal production, respectively. The 4G10 antibody was from Millipore (#05-321MG). -

TMPRSS2 and Furin Are Both Essential for Proteolytic Activation of SARS-Cov-2 in Human Airway Cells
Research Article TMPRSS2 and furin are both essential for proteolytic activation of SARS-CoV-2 in human airway cells Dorothea Bestle1,*, Miriam Ruth Heindl1,*, Hannah Limburg1,*, Thuy Van Lam van2 , Oliver Pilgram2, Hong Moulton3, David A Stein3 , Kornelia Hardes2,4, Markus Eickmann1,5, Olga Dolnik1,5 , Cornelius Rohde1,5, Hans-Dieter Klenk1, Wolfgang Garten1, Torsten Steinmetzer2, Eva Bottcher-Friebertsh¨ auser¨ 1 The novel emerged SARS-CoV-2 has rapidly spread around the broad range of mammalian and avian species, causing respiratory world causing acute infection of the respiratory tract (COVID-19) or enteric diseases. CoVs have a major surface protein, the spike (S) that can result in severe disease and lethality. For SARS-CoV-2 to protein, which initiates infection by receptor binding and fusion of enter cells, its surface glycoprotein spike (S) must be cleaved at the viral lipid envelope with cellular membranes. Like fusion two different sites by host cell proteases, which therefore rep- proteins of many other viruses, the S protein is activated by cellular resent potential drug targets. In the present study, we show that S proteases. Activation of CoV S is a complex process that requires can be cleaved by the proprotein convertase furin at the S1/S2 proteolytic cleavage of S at two distinct sites, S1/S2 and S29 (Fig 1), site and the transmembrane serine protease 2 (TMPRSS2) at the generating the subunits S1 and S2 that remain non-covalently S29 site. We demonstrate that TMPRSS2 is essential for activation linked (1, 2, 3). The S1 subunit contains the receptor binding do- of SARS-CoV-2 S in Calu-3 human airway epithelial cells through main, whereas the S2 subunit is membrane-anchored and harbors antisense-mediated knockdown of TMPRSS2 expression. -
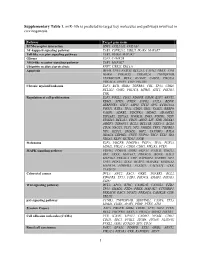
1 Supplementary Table 1. Mir-10B Is Predicted to Target Key Molecules
Supplementary Table 1. miR-10b is predicted to target key molecules and pathways involved in carcinogenesis. Pathway Target gene name ECM-receptor interaction SDC1, COL24A1, COL4A4 NF-kappa B signaling pathway TAB1, CSNK2A2, UBE2I, IRAK4, MAP3K7 Toll-like receptor signaling pathway TAB1, IRAK4, MAP3K7 Glioma E2F3, CAMK2B NOD-like receptor signaling pathway TAB1, MAP3K7 Ubiquitin mediated proteolysis RNF7, UBE2I, ERCC8 Apoptosis DFFB, TP53, FASLG, BCL2L1, CAPN2, PRKX, ATM, IRAK4, PRKACG, PRKAR2A, TNFRSF10B, TNFRSF10D, BCL2, IL1RAP, CASP8, PIK3CA, PRKACA, APAF1, CHP, PIK3R3 Chronic myeloid leukemia E2F3, BCR, GRB2, TGFBR1, CBL, TP53, CDK6, BCL2L1, GAB2, PIK3CA, MDM2, SHC1, PIK3R3, CRK Regulation of cell proliferation E2F3, FOSL2, CDX2, PDGFB, OSMR, E2F7, ARNT2, RBM5, STRN, PTEN, S1PR2, CUL3, BDNF, SERPINE1, SHC1, ASPH, ITCH, SPN, CCDC88A, FOXJ1, RXRA, TP53, CDK6, IRS1, VASH2, RBBP9, VASH1, ADRB2, PDGFRA, MDM2, ADAMTS1, EIF2AK2, EIF5A2, ICOSLG, ING5, FGFR3, NDN, ST8SIA1, BCL2L1, CDH5, ARNT, LIF, VDR, HOXA3, AGGF1, TSPAN31, BCL2, BCL11B, NKX3-1, BCL6, CD28, NACC1, FLT1, NF2, JARID2, TBX5, TGFBR1, NF1, KLF11, SMAD2, IGF2, TAX1BP3, BTLA, HDAC4, LEPRE1, CNTF, NUP62, TSC1, ETS1, ID4, NR5A2, KLF4, KCTD11, NFIB Melanoma E2F3, PDGFB, PDGFRA, FGF11, TP53, FGF23, MDM2, PIK3CA, CDK6, CDH1, PIK3R3, PTEN MAPK signaling pathway FGFR3, PDGFB, GRB2, FGF11, FASLG, GNG12, SRF, PRKX, MAP3K7, PRKACG, BDNF, RAC3, MAP3K2, PRKACA, CHP, RAPGEF2, TGFBR1, NF1, TP53, FGF23, STK4, DUSP5, MAP4K4, RPS6KA2, MAPK14, PDGFRA, PLA2G3, CACNA1C, CRK, PLA2G2F Colorectal cancer -

Cellular and Molecular Signatures in the Disease Tissue of Early
Cellular and Molecular Signatures in the Disease Tissue of Early Rheumatoid Arthritis Stratify Clinical Response to csDMARD-Therapy and Predict Radiographic Progression Frances Humby1,* Myles Lewis1,* Nandhini Ramamoorthi2, Jason Hackney3, Michael Barnes1, Michele Bombardieri1, Francesca Setiadi2, Stephen Kelly1, Fabiola Bene1, Maria di Cicco1, Sudeh Riahi1, Vidalba Rocher-Ros1, Nora Ng1, Ilias Lazorou1, Rebecca E. Hands1, Desiree van der Heijde4, Robert Landewé5, Annette van der Helm-van Mil4, Alberto Cauli6, Iain B. McInnes7, Christopher D. Buckley8, Ernest Choy9, Peter Taylor10, Michael J. Townsend2 & Costantino Pitzalis1 1Centre for Experimental Medicine and Rheumatology, William Harvey Research Institute, Barts and The London School of Medicine and Dentistry, Queen Mary University of London, Charterhouse Square, London EC1M 6BQ, UK. Departments of 2Biomarker Discovery OMNI, 3Bioinformatics and Computational Biology, Genentech Research and Early Development, South San Francisco, California 94080 USA 4Department of Rheumatology, Leiden University Medical Center, The Netherlands 5Department of Clinical Immunology & Rheumatology, Amsterdam Rheumatology & Immunology Center, Amsterdam, The Netherlands 6Rheumatology Unit, Department of Medical Sciences, Policlinico of the University of Cagliari, Cagliari, Italy 7Institute of Infection, Immunity and Inflammation, University of Glasgow, Glasgow G12 8TA, UK 8Rheumatology Research Group, Institute of Inflammation and Ageing (IIA), University of Birmingham, Birmingham B15 2WB, UK 9Institute of -

Supplementary Information Method CLEAR-CLIP. Mouse Keratinocytes
Supplementary Information Method CLEAR-CLIP. Mouse keratinocytes of the designated genotype were maintained in E-low calcium medium. Inducible cells were treated with 3 ug/ml final concentration doxycycline for 24 hours before performing CLEAR-CLIP. One 15cm dish of confluent cells was used per sample. Cells were washed once with cold PBS. 10mls of cold PBS was then added and cells were irradiated with 300mJ/cm2 UVC (254nM wavelength). Cells were then scraped from the plates in cold PBS and pelleted by centrifugation at 1,000g for 2 minutes. Pellets were frozen at -80oC until needed. Cells were then lysed on ice with occasional vortexing in 1ml of lysis buffer (50mM Tris-HCl pH 7.4, 100mM NaCl, 1mM MgCl2, 0.1 mM CaCl2, 1% NP-40, 0.5% Sodium Deoxycholate, 0.1% SDS) containing 1X protease inhibitors (Roche #88665) and RNaseOUT (Invitrogen #10777019) at 4ul/ml final concentration. Next, TurboDNase (Invitrogen #AM2238, 10U), RNase A (0.13ug) and RNase T1 (0.13U) were added and samples were incubated at 37oC for 5 minutes with occasional mixing. Samples were immediately placed on ice and then centrifuged at 16,160g at 4oC for 20 minutes to clear lysate. 25ul of Protein-G Dynabeads (Invitrogen #10004D) were used per IP. Dynabeads were pre-washed with lysis buffer and pre- incubated with 3ul of Wako Anti-Mouse-Ago2 (2D4) antibody. The dynabead/antibody mixture was added to the lysate and rocked for 2 hours at 4oC. All steps after the IP were done on bead until samples were loaded into the polyacrylamide gel. -

Under Serratia Marcescens Treatment Kai Feng1,2, Xiaoyu Lu1,2, Jian Luo1,2 & Fang Tang1,2*
www.nature.com/scientificreports OPEN SMRT sequencing of the full‑length transcriptome of Odontotermes formosanus (Shiraki) under Serratia marcescens treatment Kai Feng1,2, Xiaoyu Lu1,2, Jian Luo1,2 & Fang Tang1,2* Odontotermes formosanus (Shiraki) is an important pest in the world. Serratia marcescens have a high lethal efect on O. formosanus, but the specifc insecticidal mechanisms of S. marcescens on O. formosanus are unclear, and the immune responses of O. formosanus to S. marcescens have not been clarifed. At present, genetic database resources of O. formosanus are extremely scarce. Therefore, using O. formosanus workers infected by S. marcescens and the control as experimental materials, a full-length transcriptome was sequenced using the PacBio Sequel sequencing platform. A total of 10,364 isoforms were obtained as the fnal transcriptome. The unigenes were further annotated with the Nr, Swiss-Prot, EuKaryotic Orthologous Groups (KOG), Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) Ortholog public databases. In a comparison between the control group and a Serratia marcescens-infected group, a total of 259 diferentially expressed genes (DEGs) were identifed, including 132 upregulated and 127 downregulated genes. Pathway enrichment analysis indicated that the expression of the mitogen-activated protein kinase (MAPK) pathway, oxidative stress genes and the AMP-activated protein kinase (AMPK) pathway in O. formosanus may be associated with S. marcescens treatment. This research intensively studied O. formosanus at the high-throughput full-length transcriptome level, laying a foundation for further development of molecular markers and mining of target genes in this species and thereby promoting the biological control of O. -

Supplementary Table 2 Gene Sets Used in GSEA
Supplementary Table 2 Gene sets used in GSEA Up in RNAi and Sign Confirmed in Inducible Gene Probe Set ID Accession Symbol Gene Title 200660_at NM_005620 S100A11 S100 calcium binding protein A11 (calgizzarin) 200785_s_at NM_002332 LRP1 low density lipoprotein-related protein 1 (alpha-2-macroglobulin receptor) 201325_s_at NM_001423 EMP1 epithelial membrane protein 1 201373_at NM_000445 PLEC1 plectin 1, intermediate filament binding protein 500kDa 201466_s_at NM_002228 JUN v-jun sarcoma virus 17 oncogene homolog (avian) 201952_at AA156721 ALCAM activated leukocyte cell adhesion molecule 202042_at NM_002109 HARS histidyl-tRNA synthetase 202074_s_at NM_021980 OPTN optineurin 202087_s_at NM_001912 CTSL cathepsin L 202588_at NM_000476 AK1 adenylate kinase 1 202609_at NM_004447 EPS8 epidermal growth factor receptor pathway substrate 8 202733_at NM_004199 P4HA2 procollagen-proline, 2-oxoglutarate 4-dioxygenase (proline 4-hydroxylase), alpha polypeptide II 202756_s_at NM_002081 GPC1 glypican 1 202786_at NM_013233 STK39 serine threonine kinase 39 (STE20/SPS1 homolog, yeast) 202859_x_at NM_000584 IL8 interleukin 8 203083_at NM_003247 THBS2 thrombospondin 2 203186_s_at NM_002961 S100A4 S100 calcium binding protein A4 (calcium protein, calvasculin, metastasin, murine placental homolog) 203232_s_at NM_000332 ATXN1 ataxin 1 203233_at NM_000418 IL4R interleukin 4 receptor 203771_s_at AA740186 BLVRA biliverdin reductase A 203821_at NM_001945 HBEGF heparin-binding EGF-like growth factor 203939_at NM_002526 NT5E 5'-nucleotidase, ecto (CD73) 203955_at NM_014811 -

Coagulation Factors Directly Cleave SARS-Cov-2 Spike and Enhance Viral Entry
bioRxiv preprint doi: https://doi.org/10.1101/2021.03.31.437960; this version posted April 1, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Coagulation factors directly cleave SARS-CoV-2 spike and enhance viral entry. Edward R. Kastenhuber1, Javier A. Jaimes2, Jared L. Johnson1, Marisa Mercadante1, Frauke Muecksch3, Yiska Weisblum3, Yaron Bram4, Robert E. Schwartz4,5, Gary R. Whittaker2 and Lewis C. Cantley1,* Affiliations 1. Meyer Cancer Center, Department of Medicine, Weill Cornell Medical College, New York, NY, USA. 2. Department of Microbiology and Immunology, Cornell University, Ithaca, New York, USA. 3. Laboratory of Retrovirology, The Rockefeller University, New York, NY, USA. 4. Division of Gastroenterology and Hepatology, Department of Medicine, Weill Cornell Medicine, New York, NY, USA. 5. Department of Physiology, Biophysics and Systems Biology, Weill Cornell Medicine, New York, NY, USA. *Correspondence: [email protected] bioRxiv preprint doi: https://doi.org/10.1101/2021.03.31.437960; this version posted April 1, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Summary Coagulopathy is recognized as a significant aspect of morbidity in COVID-19 patients. The clotting cascade is propagated by a series of proteases, including factor Xa and thrombin. Other host proteases, including TMPRSS2, are recognized to be important for cleavage activation of SARS-CoV-2 spike to promote viral entry. Using biochemical and cell-based assays, we demonstrate that factor Xa and thrombin can also directly cleave SARS-CoV-2 spike, enhancing viral entry.