Investigating the Brain in Mouse Models of Duchenne Muscular Dystrophy
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

DRUGS REQUIRING PRIOR AUTHORIZATION in the MEDICAL BENEFIT Page 1
Effective Date: 08/01/2021 DRUGS REQUIRING PRIOR AUTHORIZATION IN THE MEDICAL BENEFIT Page 1 Therapeutic Category Drug Class Trade Name Generic Name HCPCS Procedure Code HCPCS Procedure Code Description Anti-infectives Antiretrovirals, HIV CABENUVA cabotegravir-rilpivirine C9077 Injection, cabotegravir and rilpivirine, 2mg/3mg Antithrombotic Agents von Willebrand Factor-Directed Antibody CABLIVI caplacizumab-yhdp C9047 Injection, caplacizumab-yhdp, 1 mg Cardiology Antilipemic EVKEEZA evinacumab-dgnb C9079 Injection, evinacumab-dgnb, 5 mg Cardiology Hemostatic Agent BERINERT c1 esterase J0597 Injection, C1 esterase inhibitor (human), Berinert, 10 units Cardiology Hemostatic Agent CINRYZE c1 esterase J0598 Injection, C1 esterase inhibitor (human), Cinryze, 10 units Cardiology Hemostatic Agent FIRAZYR icatibant J1744 Injection, icatibant, 1 mg Cardiology Hemostatic Agent HAEGARDA c1 esterase J0599 Injection, C1 esterase inhibitor (human), (Haegarda), 10 units Cardiology Hemostatic Agent ICATIBANT (generic) icatibant J1744 Injection, icatibant, 1 mg Cardiology Hemostatic Agent KALBITOR ecallantide J1290 Injection, ecallantide, 1 mg Cardiology Hemostatic Agent RUCONEST c1 esterase J0596 Injection, C1 esterase inhibitor (recombinant), Ruconest, 10 units Injection, lanadelumab-flyo, 1 mg (code may be used for Medicare when drug administered under Cardiology Hemostatic Agent TAKHZYRO lanadelumab-flyo J0593 direct supervision of a physician, not for use when drug is self-administered) Cardiology Pulmonary Arterial Hypertension EPOPROSTENOL (generic) -

Prescription Drugs Requiring Prior Authorization
PRESCRIPTION DRUGS REQUIRING PRIOR AUTHORIZATION Revised 10/16 As part of our drug utilization management program, members must request and receive prior authorization for certain prescription drugs in order to use their prescription drug benefits. Below is a list of drugs that currently require prior authorization. This list will be updated periodically as new drugs that require prior authorization are introduced. As benefits may vary by group and individual plans, the inclusion of a medication on this list does not imply prescription drug coverage. The Schedule of Benefits contains a list of drug categories that require prior authorization. Prior authorization requests are processed by our pharmacy benefit manager, Express Scripts, Inc. (ESI). Physicians must call ESI to obtain an authorization. (1-800-842-2015). Drug Name Generic Name Drug Classification Abstral fentanyl citrate oral tablet Controlled Dangerous Substance Accu-Chek Test Strips blood glucose test strips Blood Glucose Test Strips Actemra tocilizumab Monoclonal Antibody Acthar corticotropin Hormone Actimmune interferon gamma 1b Interferon Actiq fentanyl citrate OTFC Controlled Dangerous Substance Adcirca tadalafil Pulmonary Vasodilator Adempas riociguat Pulmonary Vasodilator Adlyxin lixisenatide Type 2 Diabetes Advocate Test Strips blood glucose test strips Blood Glucose Test Strips Aerospan** flunisolide Corticosteroids (Inhaled) Afrezza insulin Insulin (inhaled) Ampyra dalfampridine Multiple Sclerosis Agent Altoprev** lovastatin Cholesterol Alvesco** ciclesonide Corticosteroids -

The Use of Ataluren in the Effective Management of Duchenne Muscular Dystrophy
Review Neuromuscular Diseases Early Diagnosis and Treatment – The Use of Ataluren in the Effective Management of Duchenne Muscular Dystrophy Eugenio Mercuri,1 Ros Quinlivan2 and Sylvie Tuffery-Giraud3 1. Catholic University, Rome, Italy; 2. Great Ormond Street Hospital and National Hospital for Neurology and Neurosurgery, London, UK; 3. Laboratory of Genetics of Rare Diseases (LGMR), University of Montpellier, Montpellier, France DOI: https://doi.org/10.17925/ENR.2018.13.1.31 he understanding of the natural history of Duchenne muscular dystrophy (DMD) is increasing rapidly and new treatments are emerging that have the potential to substantially improve the prognosis for patients with this disabling and life-shortening disease. For many, Thowever, there is a long delay between the appearance of symptoms and DMD diagnosis, which reduces the possibility of successful treatment. DMD results from mutations in the large dystrophin gene of which one-third are de novo mutations and two-thirds are inherited from a female carrier. Roughly 75% of mutations are large rearrangements and 25% are point mutations. Certain deletions and nonsense mutations can be treated whereas many other mutations cannot currently be treated. This emphasises the need for early genetic testing to identify the mutation, guide treatment and inform genetic counselling. Treatments for DMD include corticosteroids and more recently, ataluren has been approved in Europe, the first disease-modifying therapy for treating DMD caused by nonsense mutations. The use of ataluren in DMD is supported by positive results from phase IIb and phase III studies in which the treatment produced marked improvements in the 6-minute walk test, timed function tests such as the 10 m walk/run test and the 4-stair ascent/descent test compared with placebo. -

Compositions and Methods for Selective Delivery of Oligonucleotide Molecules to Specific Neuron Types
(19) TZZ ¥Z_T (11) EP 2 380 595 A1 (12) EUROPEAN PATENT APPLICATION (43) Date of publication: (51) Int Cl.: 26.10.2011 Bulletin 2011/43 A61K 47/48 (2006.01) C12N 15/11 (2006.01) A61P 25/00 (2006.01) A61K 49/00 (2006.01) (2006.01) (21) Application number: 10382087.4 A61K 51/00 (22) Date of filing: 19.04.2010 (84) Designated Contracting States: • Alvarado Urbina, Gabriel AT BE BG CH CY CZ DE DK EE ES FI FR GB GR Nepean Ontario K2G 4Z1 (CA) HR HU IE IS IT LI LT LU LV MC MK MT NL NO PL • Bortolozzi Biassoni, Analia Alejandra PT RO SE SI SK SM TR E-08036, Barcelona (ES) Designated Extension States: • Artigas Perez, Francesc AL BA ME RS E-08036, Barcelona (ES) • Vila Bover, Miquel (71) Applicant: Nlife Therapeutics S.L. 15006 La Coruna (ES) E-08035, Barcelona (ES) (72) Inventors: (74) Representative: ABG Patentes, S.L. • Montefeltro, Andrés Pablo Avenida de Burgos 16D E-08014, Barcelon (ES) Edificio Euromor 28036 Madrid (ES) (54) Compositions and methods for selective delivery of oligonucleotide molecules to specific neuron types (57) The invention provides a conjugate comprising nucleuc acid toi cell of interests and thus, for the treat- (i) a nucleic acid which is complementary to a target nu- ment of diseases which require a down-regulation of the cleic acid sequence and which expression prevents or protein encoded by the target nucleic acid as well as for reduces expression of the target nucleic acid and (ii) a the delivery of contrast agents to the cells for diagnostic selectivity agent which is capable of binding with high purposes. -

Animal Models of Duchenne Muscular Dystrophy: from Basic Mechanisms to Gene Therapy Joe W
© 2015. Published by The Company of Biologists Ltd | Disease Models & Mechanisms (2015) 8, 195-213 doi:10.1242/dmm.018424 REVIEW Animal models of Duchenne muscular dystrophy: from basic mechanisms to gene therapy Joe W. McGreevy1, Chady H. Hakim1, Mark A. McIntosh1 and Dongsheng Duan1,2,* ABSTRACT The identification of the disease-causing gene and the molecular Duchenne muscular dystrophy (DMD) is a progressive muscle- basis for the DMD and BMD phenotypes establishes the foundation wasting disorder. It is caused by loss-of-function mutations in the for DMD gene therapy (Fig. 2A). To mitigate muscle disease, one dystrophin gene. Currently, there is no cure. A highly promising can either restore the full-length transcript or express a truncated but therapeutic strategy is to replace or repair the defective dystrophin in-frame dystrophin gene (Duan, 2011; Goyenvalle et al., 2011; gene by gene therapy. Numerous animal models of DMD have been Konieczny et al., 2013; Mendell et al., 2012; Verhaart and Aartsma- developed over the last 30 years, ranging from invertebrate to large Rus, 2012). Several gene therapy strategies are currently under mammalian models. mdx mice are the most commonly employed development. They include replacing the mutated gene with a models in DMD research and have been used to lay the groundwork functional candidate gene (gene replacement) or repairing the for DMD gene therapy. After ~30 years of development, the field has defective gene by targeted correction and exon skipping (gene reached the stage at which the results in mdx mice can be validated repair). Currently, adeno-associated virus (AAV)-mediated gene and scaled-up in symptomatic large animals. -
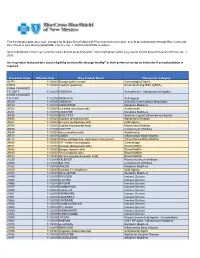
Procedure Code Effective Date Drug Product Name* Therapeutic
This list includes procedure code changes for Medical Benefit Specialty Pharmacy that may require benefit preauthorization through Blue Cross and Blue Shield of New Mexico (BCBSNM) effective Jan. 1, 2020 for BCBSNM members. Items highlighted in blue may currently require benefit preauthorization. Items highlighted yellow may require benefit preauthorization effective Jan. 1, 2020. It is imperative that providers check eligibility and benefits through Availity® or their preferred vendor to determine if preauthorization is required. Procedure Code Effective Date Drug Product Name* Therapeutic Category 90378 1/1/2020 Synagis (palivizumab) Immunological Agent C9036 1/1/2020 Onpattro (patisiran) Small interfering RNA (siRNA) C9466 CHANGED TO J0517 1/1/2019 FASENRA Antiasthmatic - Monoclonal Antibodies C9493 CHANGED TO J1301 1/1/2019 RADICAVA ALS Agents J0129 1/1/2019 ORENCIA Selective Costimulation Modulators J0180 1/1/2019 FABRAZYME Metabolic Modifiers J0202 1/1/2020 Lemtrada (alemtuzumab) Autoimmune J0221 1/1/2019 LUMIZYME Metabolic Modifiers J0490 1/1/2019 BENLYSTA Systemic Lupus Erythematosus Agents J0565 1/1/2020 Zinplava (bezlotoxumab) Monoclonal Antibody J0567 1/1/2020 Brineura (cerliponase alfa) CNS Agents J0584 1/1/2020 Crysvita (burosumab-twza) Monoclonal Antibody J0598 1/1/2019 CINRYZE Complement Inhibitors J0638 1/1/2020 Ilaris (canakinumab) Autoimmune J0717 1/1/2019 CIMZIA Inflammatory Bowel Agents J0775 1/1/2020 Xiaflex (collagenase, clostridium histolyticum) Tissue Permeability Modifier J0800 1/1/2020 H.P. Acthar (corticotropin) -

Glia Fiziológia Gliális Neurotranszmitter Transzporterek
Glia fiziológia Gliális neurotranszmitter transzporterek Glutamát transzporterek asztrocitákban EAA- Excitatory Amino Acid Transporter (EAAT) család GLAST asztroglia, radiális glián (EAAT1) GLT1 (EAAT2) főleg asztroglia, de bizonyos neuronokon is EAAC1 (EAAT3) érett neuronok EAAT4 Purkinje sejtek, GABA-erg interneuronok, retina EAAT5 retina bipoláris és fotoreceptor sejtjei GLAST: Glutamate Aspartate Transporter GLT1: Glutamate Transporter 1 EAAC1: Excitatory Amino Acid Carrier 1 [Glu]EC: 2-5 uM (ez megnő persze neuron-tüzeléskor) [Glu]IC: 1-10 mM tehát Glu eltávolítás nagy koncentráció-gradienssel szemben kell, hogy történjen ! („uphill” translocation) Glia fiziológia Gliális neurotranszmitter transzporterek Glutamát transzporterek asztrocitákban be: 1 Glu-, 3 Na+, 1H+ glia ki: 1 K+ ionok koncentráció- gradiensük szerint mozognak alacsony Na+ic fenntartása kritikus a Glutamát elektrogén a transzporter mert uptake szempontjából !!! befelé irányuló áram van és deploarizáció: a benti + töltés tovább segíti a – glutamát felvételét Glia fiziológia Gliális neurotranszmitter transzporterek Glutamát transzporterek asztrocitákban Glutamát: sokféle ionmozgást okoz asztroban be: 1Glu- AMPAR aktiváció: 3Na+, 1H+ be: Na+ ki: K+ 1. Na+/K+ pumpa eredmény: net Na+ influx 2. Na+/Ca++ exchanger Na+ ~5mM 20-30 mM-ra nő ki: Na+ be: K+ ic (NCX) gyors megfordulása: energiaigényes és ki: Na+ be: Ca++ !!! lassabb ezt ellensúlyozza 1. és 2. Glia fiziológia Gliális neurotranszmitter transzporterek Glutamát transzporterek asztrocitákban alacsony Na+ic fenntartása -

Sorting of the Vesicular GABA Transporter to Functional Vesicle Pools by an Atypical Dileucine-Like Motif
10634 • The Journal of Neuroscience, June 26, 2013 • 33(26):10634–10646 Cellular/Molecular Sorting of the Vesicular GABA Transporter to Functional Vesicle Pools by an Atypical Dileucine-like Motif Magda S. Santos,1 C. Kevin Park,1 Sarah M. Foss,1,2 Haiyan Li,1 and Susan M. Voglmaier1 1Department of Psychiatry, and 2Graduate Program in Cell Biology, University of California, San Francisco, School of Medicine, San Francisco, California 94143-0984 Increasing evidence indicates that individual synaptic vesicle proteins may use different signals, endocytic adaptors, and trafficking pathways for sorting to distinct pools of synaptic vesicles. Here, we report the identification of a unique amino acid motif in the vesicular GABA transporter (VGAT) that controls its synaptic localization and activity-dependent recycling. Mutational analysis of this atypical dileucine-like motif in rat VGAT indicates that the transporter recycles by interacting with the clathrin adaptor protein AP-2. However, mutation of a single acidic residue upstream of the dileucine-like motif leads to a shift to an AP-3-dependent trafficking pathway that preferentially targets the transporter to the readily releasable and recycling pool of vesicles. Real-time imaging with a VGAT-pHluorin fusion provides a useful approach to explore how unique sorting sequences target individual proteins to synaptic vesicles with distinct functional properties. Introduction ery to different vesicle pools, or molecular heterogeneity of SVs How proteins are sorted to synaptic vesicles (SVs) has been a that could determine their functional characteristics (Mor- long-standing question in cell biology. At the nerve terminal, genthaler et al., 2003; Salazar et al., 2004; Voglmaier and Ed- synaptic vesicles undergo exocytosis and then reform through wards, 2007; Hua et al., 2011a; Lavoie et al., 2011; Raingo et al., endocytic events. -

Med Pharm PA List to Build Pdfs.Xlsx
The following medications, typically administered in a provider's office or ambulatory/outpatient infusion center, require authorization prior to utilization. Authorization requests can be submitted via the Portal. Additionally, you are welcome to call our member services line to initiate the Prior Authorization process. This list is updated quaterly. Effective date: 1/1/2021 Therapeutic category Brand Name Generic Name HCPCS Immunologic agent Orencia, IV infusion abatacept J0129 Botulinum toxins Dysport abobotulinumtoxin B J0586 HER-2 receptor drug Kadcyla ado-trastuzumab emtansine J9354 Ophthalmic injectable Eylea afilbercept J0178 Enzyme replacement drug Fabrazyme agalsidase beta J0180 Multiple sclerosis drug Lemtrada alemtuzumab J0202 Enzyme replacement drug Lumizyme alglucosidase alfa J0221 Enzyme replacement drug Strensiq asfotase alfa J3590 (non-specific) PD1-PDL1 drug Tecentriq atezolizumab J9022 PD1-PDL1 drug Bavencio avelumab J9023 CAR-T Therapy Yescarta axicabtageneciloleucel inpatient admin Systemic lupus erythematosus (SLE) drug Benlysta belimumab J0490 Antineoplastic agent Beleodaq belinostat J9032 Antineoplastic Belrapzo bendamustine HCl J9036 Antineoplastic Bendeka bendamustine HCl J9034 Antineoplastic agent Treanda bendamustine HCl J9033 Respiratory injectable Fasenra benralizumab J0517 J9035/J3590 (ophthalmology Antineoplastic Avastin bevacizumab uses this non-specific code) Antineoplastic agent Mvasi bevacizumab-awwb Q5107 Antineoplastic agent Zirabev bevacizumab-bvzr Q5118 Antineoplastic agent Blincyto blinatumomab -

1997 Mcintire UNC-47.Pdf
letters to nature murine Jnk1 cDNA. To test c-jun and c-fos expression, a 199-bp fragment 18. Ferkany, J. W., Zaczek, R. & Coyle, J. T. The mechanism of kainic acid neurotoxicity. Nature 308, 561– 562 (1984). corresponding to nucleotides 891–1,089 of the murine c-jun cDNA and a 346-bp 19. Morgan, J. I. & Curran, T. Stimulus-transcription coupling in the nervous system: involvement of the fragment corresponding to nucleotides 2,173–2,518 of the murine c-fos cDNA inducible proto-oncogenes fos and jun. Annu. Rev. Neurosci. 14, 421–451 (1991). were used for the generation of radiolabelled probes for northern hybridization 20. Kasof, G. M. et al. Kanic acid-induced neuronal death is associated with DNA damage and a unique immediate-early gene response in c-fos-lacZ transgenic rats. J. Neurosci. 15, 4238–4249 (1995). analysis. JNK activity in hippocampal lysates (30 mg) was measured before and 21. Morgan, J. I., Cohen, D. R., Hempstead, J. L. & Curran, T. Mapping patterns of c-fos expression in the after immunodepletion of Jnk1 and Jnk2 by in-gel protein kinase assays using central nervous system after seizure. Science 237, 192–197 (1987). 3 9 22. Berger, M. & Ben-Ari, Y. Autoradiographic visualization of [ H]kainic acid receptor subtypes in the the substrate glutathione S-transferase (GST)–cJun . rat hippocampus. Neurosci. Lett. 39, 237–242 (1983). Luciferase activity assay. Mice were decapitated and the brains dissected. 23. Westbrook, G. L. & Lothman, E. W. Cellular and synaptic basis of kainic acid-induced hippocampal Brain tissues were immediately lysed (Promega) and the luciferase activity was epileptiform activity. -

Deflazacort, Eteplirsen, and Golodirsen for Duchenne Muscular Dystrophy: Effectiveness and Value
Deflazacort, Eteplirsen, and Golodirsen for Duchenne Muscular Dystrophy: Effectiveness and Value Final Evidence Report August 15, 2019 Prepared for ©Institute for Clinical and Economic Review, 2019 University of Illinois at Chicago College of Pharmacy ICER Staff and Consultants Modeling Group Grace A. Lin, MD Catherine Koola, MPH Surrey M. Walton, PhD Associate Professor of Program Manager Associate Professor, Pharmacy Systems, Outcomes and Policy Medicine and Health Policy ICER Assistant Director, Center for Pharmacoepidemiology and University of California, San Pharmacoeconomic Research Francisco University of Illinois at Chicago College of Pharmacy Foluso Agboola, MBBS, MPH Matt Seidner Nicole Boyer, PhD Director, Evidence Synthesis Program Director Postdoctoral Fellow ICER ICER The University of Chicago Noemi Fluetsch, MPH Rick Chapman, PhD, MS Danny Quach, PharmD Research Assistant, Health Director of Health Economics University of Illinois at Chicago College of Pharmacy Economics and Outcomes ICER Research ICER Varun M. Kumar, MBBS, MPH, David Rind, MD, MSc MSc Chief Medical Officer Associate Director of Health ICER Economics ICER The role of the University of Illinois (UIC) College of Pharmacy Sumeyye Samur, PhD, MS Steve Pearson, MD, MSc Modeling Group is limited to the development of the cost- Health Economist President effectiveness model, and the resulting ICER reports do not ICER ICER necessarily represent the views of the UIC. DATE OF PUBLICATION: August 15, 2019 Grace Lin served as the lead author for the report and wrote the background, other benefits, and contextual considerations sections of the report. Foluso Agboola was responsible for the oversight of the systematic review and authorship of the comparative clinical effectiveness section with the support of Ifeoma Otuonye and Noemi Fluetsch. -

What Is the Level of Dystrophin Expression Required for Effective
RVC OPEN ACCESS REPOSITORY – COPYRIGHT NOTICE This is the author’s accepted manuscript. The version of record is available online via the Journal of Muscle Research and Cell Motility: https://doi.org/10.1007/s10974-019-09535-9. The full details of the published version of the article are as follows: TITLE: What is the level of dystrophin expression required for effective therapy of Duchenne muscular dystrophy? AUTHORS: DJ Wells JOURNAL TITLE: Journal of Muscle Research and Cell Motility PUBLICATION DATE: 9 July 2019 PUBLISHER: Springer Nature DOI: 10.1007/s10974-019-09535-9 What is the level of dystrophin expression required for effective therapy of Duchenne muscular dystrophy? Dominic J. Wells. Neuromuscular Diseases Group, Department of Comparative Biomedical Sciences, Royal Veterinary College, Royal College Street NW1 0TU, UK. [email protected] orcid.org/0000-0002-1425-6344 Abstract Duchenne muscular dystrophy (DMD) is a fatal X-linked muscle wasting disease. The disease is due to mutations in the DMD gene that encodes for a large intracellular protein called dystrophin. Dystrophin plays a critical role in linking the internal cytoskeleton of the striated muscle cell with the extracellular matrix as well as having cell signalling functions. In its absence muscle contraction is associated with cycles of damage, repair, inflammation and fibrosis with eventual loss of muscle and replacement with fat. Experiments in animal models of DMD have generated a number of different approaches to the induction of dystrophin including viral vector mediated delivery of a recombinant dystrophin gene, antisense oligonucleotide mediated exon-skipping to restore the open reading frame in the dystrophin mRNA, read-through of premature stop mutations, genome modification using CRISPR-Cas9 or cell based transfer of a functional dystrophin gene.