The Use of Ataluren in the Effective Management of Duchenne Muscular Dystrophy
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

2017 ANNUAL REPORT | Translating SCIENCE • Transforming LIVES OUR COMMITMENT Make Every Day Count at PTC, Patients Are at the Center of Everything We Do
20 YEARS OF COMMITMENT 2017 ANNUAL REPORT | Translating SCIENCE • Transforming LIVES OUR COMMITMENT Make every day count At PTC, patients are at the center of everything we do. We have the opportunity to support patients and families living with rare disorders through their journey. We know that every day matters and we are committed to making a difference. OUR SCIENCE Our scientists are finding new ways to regulate biology to control disease We have several scientific research platforms focused on modulating protein expression within the cell that we believe have the potential to address many rare genetic disorders. OUR PEOPLE Care for each other, our community, and for the needs of our patients At PTC, we are looking at drug discovery and development in a whole new light, bringing new technologies and approaches to developing medicines for patients living with rare disorders and cancer. We strive every day to be better than we were the day before. At PTC Therapeutics, it is our mission to provide access to best-in-class treatments for patients who have an unmet need. We are a science-led, global biopharmaceutical company focused on the discovery, development and commercialization of clinically-differentiated medicines that provide benefits to patients with rare disorders. Founded 20 years ago, PTC Therapeutics has successfully launched two rare disorder products and has a global commercial footprint. This success is the foundation that drives investment in a robust pipeline of transformative medicines and our mission to provide access to best-in-class treatments for patients who have an unmet medical need. As we celebrate our 20th year of bringing innovative therapies to patients affected by rare disorders, we reflect on our unwavering commitment to our patients, our science and our employees. -

DRUGS REQUIRING PRIOR AUTHORIZATION in the MEDICAL BENEFIT Page 1
Effective Date: 08/01/2021 DRUGS REQUIRING PRIOR AUTHORIZATION IN THE MEDICAL BENEFIT Page 1 Therapeutic Category Drug Class Trade Name Generic Name HCPCS Procedure Code HCPCS Procedure Code Description Anti-infectives Antiretrovirals, HIV CABENUVA cabotegravir-rilpivirine C9077 Injection, cabotegravir and rilpivirine, 2mg/3mg Antithrombotic Agents von Willebrand Factor-Directed Antibody CABLIVI caplacizumab-yhdp C9047 Injection, caplacizumab-yhdp, 1 mg Cardiology Antilipemic EVKEEZA evinacumab-dgnb C9079 Injection, evinacumab-dgnb, 5 mg Cardiology Hemostatic Agent BERINERT c1 esterase J0597 Injection, C1 esterase inhibitor (human), Berinert, 10 units Cardiology Hemostatic Agent CINRYZE c1 esterase J0598 Injection, C1 esterase inhibitor (human), Cinryze, 10 units Cardiology Hemostatic Agent FIRAZYR icatibant J1744 Injection, icatibant, 1 mg Cardiology Hemostatic Agent HAEGARDA c1 esterase J0599 Injection, C1 esterase inhibitor (human), (Haegarda), 10 units Cardiology Hemostatic Agent ICATIBANT (generic) icatibant J1744 Injection, icatibant, 1 mg Cardiology Hemostatic Agent KALBITOR ecallantide J1290 Injection, ecallantide, 1 mg Cardiology Hemostatic Agent RUCONEST c1 esterase J0596 Injection, C1 esterase inhibitor (recombinant), Ruconest, 10 units Injection, lanadelumab-flyo, 1 mg (code may be used for Medicare when drug administered under Cardiology Hemostatic Agent TAKHZYRO lanadelumab-flyo J0593 direct supervision of a physician, not for use when drug is self-administered) Cardiology Pulmonary Arterial Hypertension EPOPROSTENOL (generic) -

Opportunities and Challenges for Antisense Oligonucleotide Therapies
Received: 10 January 2020 Revised: 23 April 2020 Accepted: 8 May 2020 DOI: 10.1002/jimd.12251 REVIEW ARTICLE Opportunities and challenges for antisense oligonucleotide therapies Elsa C. Kuijper1 | Atze J. Bergsma2,3 | W.W.M. Pim Pijnappel2,3 | Annemieke Aartsma-Rus1 1Department of Human Genetics, Leiden University Medical Center, Leiden, The Abstract Netherlands Antisense oligonucleotide (AON) therapies involve short strands of modi- 2Department of Pediatrics, Center for fied nucleotides that target RNA in a sequence-specific manner, inducing Lysosomal and Metabolic Diseases, targeted protein knockdown or restoration. Currently, 10 AON therapies Erasmus Medical Center, Rotterdam, The Netherlands have been approved in the United States and Europe. Nucleotides are chem- 3Department of Clinical Genetics, Center ically modified to protect AONs from degradation, enhance bioavailability for Lysosomal and Metabolic Diseases, and increase RNA affinity. Whereas single stranded AONs can efficiently Erasmus Medical Center, Rotterdam, The Netherlands be delivered systemically, delivery of double stranded AONs requires capsulation in lipid nanoparticles or binding to a conjugate as the uptake Correspondence enhancing backbone is hidden in this conformation. With improved chem- Annemieke Aartsma-Rus, LUMC Postzone S4-P, Albinusdreef 2, 2333 ZA istry, delivery vehicles and conjugates, doses can be lowered, thereby reduc- Leiden, The Netherlands. ing the risk and occurrence of side effects. AONs can be used to knockdown Email: [email protected] or restore levels of protein. Knockdown can be achieved by single stranded Communicating Editor: Carla E. Hollak or double stranded AONs binding the RNA transcript and activating RNaseH-mediated and RISC-mediated degradation respectively. Transcript binding by AONs can also prevent translation, hence reducing protein levels. -

Duchenne Muscular Dystrophy (DMD) Agents
Therapeutic Class Overview Duchenne muscular dystrophy (DMD) Agents INTRODUCTION • Duchenne muscular dystrophy (DMD) is 1 of 4 conditions known as dystrophinopathies, which are inherited, X-linked myopathic disorders due to a defect in the dystrophin gene that results in the primary pathologic process of muscle fiber degradation. The hallmark symptom is progressive weakness (Darras 2018[a], Darras 2018[b], Muscular Dystrophy Association [MDA] 2019). The other 3 conditions include: Becker muscular dystrophy (BMD), which is a mild form of DMD; an intermediate presentation between BMD and DMD; and DMD-associated dilated cardiomyopathy, which has little or no clinical ○ skeletal or muscle disease (MDA 2019). • DMD symptom onset is in early childhood, usually between the ages of 2 and 3 years old. The proximal muscles are affected first, followed by the distal limb muscles. Generally, the lower external muscles will be affected before the upper. The affected child may have difficulties jumping, walking, and running (MDA 2019). • The prevalence of DMD ranges from 1 to 2 per 10,000 live male births; female-manifesting carriers are rarer, but can present with a range of symptoms that vary in their severities (Birnkrant et al 2018, Darras 2018[a], Emflaza Food and Drug Administration [FDA] Medical Review 2017). • The clinical course and lifespan of patients with DMD is relatively short. Individuals are usually confined to a wheelchair by age 13, and many die in their late teens or twenties from respiratory insufficiency or cardiomyopathy. Although survival until adulthood is more common now, very few patients survive past the 3rd decade (Darras 2018[a]). -

Prescription Drugs Requiring Prior Authorization
PRESCRIPTION DRUGS REQUIRING PRIOR AUTHORIZATION Revised 10/16 As part of our drug utilization management program, members must request and receive prior authorization for certain prescription drugs in order to use their prescription drug benefits. Below is a list of drugs that currently require prior authorization. This list will be updated periodically as new drugs that require prior authorization are introduced. As benefits may vary by group and individual plans, the inclusion of a medication on this list does not imply prescription drug coverage. The Schedule of Benefits contains a list of drug categories that require prior authorization. Prior authorization requests are processed by our pharmacy benefit manager, Express Scripts, Inc. (ESI). Physicians must call ESI to obtain an authorization. (1-800-842-2015). Drug Name Generic Name Drug Classification Abstral fentanyl citrate oral tablet Controlled Dangerous Substance Accu-Chek Test Strips blood glucose test strips Blood Glucose Test Strips Actemra tocilizumab Monoclonal Antibody Acthar corticotropin Hormone Actimmune interferon gamma 1b Interferon Actiq fentanyl citrate OTFC Controlled Dangerous Substance Adcirca tadalafil Pulmonary Vasodilator Adempas riociguat Pulmonary Vasodilator Adlyxin lixisenatide Type 2 Diabetes Advocate Test Strips blood glucose test strips Blood Glucose Test Strips Aerospan** flunisolide Corticosteroids (Inhaled) Afrezza insulin Insulin (inhaled) Ampyra dalfampridine Multiple Sclerosis Agent Altoprev** lovastatin Cholesterol Alvesco** ciclesonide Corticosteroids -

Circulating Biomarkers in Neuromuscular Disorders: What Is Known, What Is New
biomolecules Review Circulating Biomarkers in Neuromuscular Disorders: What Is Known, What Is New Andrea Barp 1,* , Amanda Ferrero 1, Silvia Casagrande 1,2 , Roberta Morini 1 and Riccardo Zuccarino 1 1 NeuroMuscular Omnicentre (NeMO) Trento, Villa Rosa Hospital, Via Spolverine 84, 38057 Pergine Valsugana, Italy; [email protected] (A.F.); [email protected] (S.C.); [email protected] (R.M.); [email protected] (R.Z.) 2 Department of Neurosciences, Drug and Child Health, University of Florence, Largo Brambilla 3, 50134 Florence, Italy * Correspondence: [email protected] Abstract: The urgent need for new therapies for some devastating neuromuscular diseases (NMDs), such as Duchenne muscular dystrophy or amyotrophic lateral sclerosis, has led to an intense search for new potential biomarkers. Biomarkers can be classified based on their clinical value into different categories: diagnostic biomarkers confirm the presence of a specific disease, prognostic biomarkers provide information about disease course, and therapeutic biomarkers are designed to predict or measure treatment response. Circulating biomarkers, as opposed to instrumental/invasive ones (e.g., muscle MRI or nerve ultrasound, muscle or nerve biopsy), are generally easier to access and less “time-consuming”. In addition to well-known creatine kinase, other promising molecules seem to be candidate biomarkers to improve the diagnosis, prognosis and prediction of therapeutic response, such as antibodies, neurofilaments, and microRNAs. However, there are some criticalities that can complicate their application: variability during the day, stability, and reliable performance metrics Citation: Barp, A.; Ferrero, A.; (e.g., accuracy, precision and reproducibility) across laboratories. In the present review, we discuss Casagrande, S.; Morini, R.; Zuccarino, the application of biochemical biomarkers (both validated and emerging) in the most common NMDs R. -

Are Speci C Symptoms and Signs That Will Enable the Clinician to Recognise Disease Patterns That Will Narrow Down the Differenti
CHILDHOOD DEVELOPMENTAL SCREENING 2020 https://doi.org/10.33591/sfp.46.5.u5 UNIT NO. 5 Brumbaugh D, Case LE, Clemens PR, Hadjiyannakis S, Pandya S, Street CHILDHOOD NEUROMUSCULAR DISORDERS examiner’s hands under the armpits and assessed for any arm periodic paralysis) and the rate of progression. An acute or delay/hypotonia, neuroimaging with MRI brain and aordable gene panels targeting neuromuscular disorders promising, with near-normal motor development in some receptor but reduced transcriptional activation and therefore 3. Orthopaedic Care neuromuscular diseases has resulted in faster and more accurate slip sign, and an attempt should be made to see if the infant can subacute onset of symptoms may be suggestive of an endocrine appropriate chromosomal/genetic testing can be considered. which are faster and less costly than whole exome or whole infants. is has resulted in new recommendations for neonatal has less side eects of other steroids. Clinical trials are still Prevention of contractures may be aected with appropriate diagnosis. Furthermore, new treatments are in the pipeline or N. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointes- A/Prof Stacey Tay Kiat Hong bear weight on both legs. e infant is then put into ventral or inammatory myositis, the chronic onset of symptoms may 2. If the child has evidence of lower limb involvement and genome sequencing. e choice of whole exome or whole newborn screening of SMA as well as a treatment algorithm for underway for head-to-head comparison of Vamorolone and resting splints of the ankles. Proper seating is also key to are already available for certain neuromuscular disorders, and 11 20 tinal and nutritional management. -

For Cystic Fibrosis (Review)
Cochrane Database of Systematic Reviews Ataluren and similar compounds (specific therapies for premature termination codon class I mutations) for cystic fibrosis (Review) Aslam AA, Higgins C, Sinha IP, Southern KW Aslam AA, Higgins C, Sinha IP, Southern KW. Ataluren and similar compounds (specific therapies for premature termination codon class I mutations) for cystic fibrosis. Cochrane Database of Systematic Reviews 2017, Issue 1. Art. No.: CD012040. DOI: 10.1002/14651858.CD012040.pub2. www.cochranelibrary.com Ataluren and similar compounds (specific therapies for premature termination codon class I mutations) for cystic fibrosis (Review) Copyright © 2017 The Cochrane Collaboration. Published by John Wiley & Sons, Ltd. TABLE OF CONTENTS HEADER....................................... 1 ABSTRACT ...................................... 1 PLAINLANGUAGESUMMARY . 2 SUMMARY OF FINDINGS FOR THE MAIN COMPARISON . ..... 4 BACKGROUND .................................... 7 OBJECTIVES ..................................... 8 METHODS ...................................... 8 RESULTS....................................... 12 Figure1. ..................................... 13 Figure2. ..................................... 16 DISCUSSION ..................................... 20 AUTHORS’CONCLUSIONS . 22 ACKNOWLEDGEMENTS . 22 REFERENCES ..................................... 23 CHARACTERISTICSOFSTUDIES . 26 DATAANDANALYSES. 33 Analysis 1.1. Comparison 1 Ataluren versus placebo, Outcome 1 FEV - mean relative change from baseline. 35 Analysis 1.2. Comparison 1 -

Ataluren Stimulates Ribosomal Selection of Near-Cognate Trnas to Promote Nonsense Suppression
Ataluren stimulates ribosomal selection of near-cognate tRNAs to promote nonsense suppression Bijoyita Roya,b,1, Westley J. Friesenb,1, Yuki Tomizawab, John D. Leszykc, Jin Zhuob, Briana Johnsonb, Jumana Dakkab, Christopher R. Trottab, Xiaojiao Xueb,d,e, Venkateshwar Mutyame,f, Kim M. Keelingd,e, James A. Mobleyg, Steven M. Rowee,f, David M. Bedwelld,e, Ellen M. Welchb, and Allan Jacobsona,2 aDepartment of Microbiology and Physiological Systems, University of Massachusetts Medical School, Worcester, MA 01655-0122; bPTC Therapeutics Inc., South Plainfield, NJ 07080; cDepartment of Biochemistry and Molecular Pharmacology, University of Massachusetts Medical School, Worcester, MA 01655-0122; dDepartment of Biochemistry and Molecular Genetics, University of Alabama at Birmingham, Birmingham, AL 35294; eGregory Fleming James Cystic Fibrosis Research Center, University of Alabama at Birmingham, Birmingham, AL 35294; fDepartment of Medicine, University of Alabama at Birmingham, Birmingham, AL 35294; and gDepartment of Surgery, University of Alabama at Birmingham, Birmingham, AL 35294 Edited by Rachel Green, Johns Hopkins University, Baltimore, MD, and approved September 6, 2016 (received for review April 1, 2016) A premature termination codon (PTC) in the ORF of an mRNA promote therapeutic nonsense suppression (1, 3). To date, ataluren generally leads to production of a truncated polypeptide, accelerated has been shown to restore function to more than 20 different degradation of the mRNA, and depression of overall mRNA ex- disease-specific or reporter nonsense alleles in systems ranging in pression. Accordingly, nonsense mutations cause some of the most complexity from in vitro translation to cell culture to mouse models severe forms of inherited disorders. The small-molecule drug ataluren and human patients (1, 3–14). -

PRAC Draft Agenda of Meeting 14-17 March 2016
14 March 2016 EMA/PRAC/199507/2016 Corr.∗ Procedure Management and Committees Support Division Pharmacovigilance Risk Assessment Committee (PRAC) Draft agenda for the meeting on 14-17 March 2016 Chair: June Raine – Vice-Chair: Almath Spooner 14 March 2016, 13:00 – 19:00, room 3/A 15 March 2016, 08:30 – 19:00, room 3/A 16 March 2016, 08:30 – 19:00, room 3/A 17 March 2016, 08:30 – 16:00, room 3/E Organisational, regulatory and methodological matters (ORGAM) 31 March 2016, 10:00 - 12:00, room 7/B, via Adobe Connect Health and safety information In accordance with the Agency’s health and safety policy, delegates are to be briefed on health, safety and emergency information and procedures prior to the start of the meeting. Disclaimers Some of the information contained in this agenda is considered commercially confidential or sensitive and therefore not disclosed. With regard to intended therapeutic indications or procedure scopes listed against products, it must be noted that these may not reflect the full wording proposed by applicants and may also change during the course of the review. Additional details on some of these procedures will be published in the PRAC meeting highlights once the procedures are finalised. Of note, this agenda is a working document primarily designed for PRAC members and the work the Committee undertakes. Note on access to documents Some documents mentioned in the agenda cannot be released at present following a request for access to documents within the framework of Regulation (EC) No 1049/2001 as they are subject to on- going procedures for which a final decision has not yet been adopted. -

Animal Models of Duchenne Muscular Dystrophy: from Basic Mechanisms to Gene Therapy Joe W
© 2015. Published by The Company of Biologists Ltd | Disease Models & Mechanisms (2015) 8, 195-213 doi:10.1242/dmm.018424 REVIEW Animal models of Duchenne muscular dystrophy: from basic mechanisms to gene therapy Joe W. McGreevy1, Chady H. Hakim1, Mark A. McIntosh1 and Dongsheng Duan1,2,* ABSTRACT The identification of the disease-causing gene and the molecular Duchenne muscular dystrophy (DMD) is a progressive muscle- basis for the DMD and BMD phenotypes establishes the foundation wasting disorder. It is caused by loss-of-function mutations in the for DMD gene therapy (Fig. 2A). To mitigate muscle disease, one dystrophin gene. Currently, there is no cure. A highly promising can either restore the full-length transcript or express a truncated but therapeutic strategy is to replace or repair the defective dystrophin in-frame dystrophin gene (Duan, 2011; Goyenvalle et al., 2011; gene by gene therapy. Numerous animal models of DMD have been Konieczny et al., 2013; Mendell et al., 2012; Verhaart and Aartsma- developed over the last 30 years, ranging from invertebrate to large Rus, 2012). Several gene therapy strategies are currently under mammalian models. mdx mice are the most commonly employed development. They include replacing the mutated gene with a models in DMD research and have been used to lay the groundwork functional candidate gene (gene replacement) or repairing the for DMD gene therapy. After ~30 years of development, the field has defective gene by targeted correction and exon skipping (gene reached the stage at which the results in mdx mice can be validated repair). Currently, adeno-associated virus (AAV)-mediated gene and scaled-up in symptomatic large animals. -
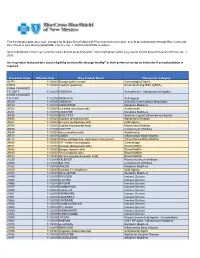
Procedure Code Effective Date Drug Product Name* Therapeutic
This list includes procedure code changes for Medical Benefit Specialty Pharmacy that may require benefit preauthorization through Blue Cross and Blue Shield of New Mexico (BCBSNM) effective Jan. 1, 2020 for BCBSNM members. Items highlighted in blue may currently require benefit preauthorization. Items highlighted yellow may require benefit preauthorization effective Jan. 1, 2020. It is imperative that providers check eligibility and benefits through Availity® or their preferred vendor to determine if preauthorization is required. Procedure Code Effective Date Drug Product Name* Therapeutic Category 90378 1/1/2020 Synagis (palivizumab) Immunological Agent C9036 1/1/2020 Onpattro (patisiran) Small interfering RNA (siRNA) C9466 CHANGED TO J0517 1/1/2019 FASENRA Antiasthmatic - Monoclonal Antibodies C9493 CHANGED TO J1301 1/1/2019 RADICAVA ALS Agents J0129 1/1/2019 ORENCIA Selective Costimulation Modulators J0180 1/1/2019 FABRAZYME Metabolic Modifiers J0202 1/1/2020 Lemtrada (alemtuzumab) Autoimmune J0221 1/1/2019 LUMIZYME Metabolic Modifiers J0490 1/1/2019 BENLYSTA Systemic Lupus Erythematosus Agents J0565 1/1/2020 Zinplava (bezlotoxumab) Monoclonal Antibody J0567 1/1/2020 Brineura (cerliponase alfa) CNS Agents J0584 1/1/2020 Crysvita (burosumab-twza) Monoclonal Antibody J0598 1/1/2019 CINRYZE Complement Inhibitors J0638 1/1/2020 Ilaris (canakinumab) Autoimmune J0717 1/1/2019 CIMZIA Inflammatory Bowel Agents J0775 1/1/2020 Xiaflex (collagenase, clostridium histolyticum) Tissue Permeability Modifier J0800 1/1/2020 H.P. Acthar (corticotropin)