Procedures, Programs and Drugs You Must Precertify
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Spinraza™ (Nusinersen)
Spinraza™ (nusinersen) (Intrathecal) Document Number: IC-0291 Last Review Date: 08/03/2021 Date of Origin: 01/31/2017 Dates Reviewed: 01/2017, 02/2017, 01/2018, 08/2018, 06/2019, 08/2020, 08/2021 I. Length of Authorization Coverage will be provided annually and may be renewed. II. Dosing Limits A. Quantity Limit (max daily dose) [NDC Unit]: • Loading: 1 vial on D1, D15, D29, and D59 • Maintenance: 1 vial every 112 days B. Max Units (per dose and over time) [HCPCS Unit]: • Loading: 120 billable units on D1, D15, D29, and D59 • Maintenance: 120 billable units every 112 days III. Initial Approval Criteria1-12 • Submission of medical records related to the medical necessity criteria is REQUIRED on all requests for authorizations. Records will be reviewed at the time of submission. Please provide documentation via direct upload through the PA web portal or by fax. Coverage is provided in the following conditions: Spinal Muscular Atrophy (SMA) † Ф • Patient must not have previously received treatment with SMA gene therapy (i.e., onasemnogene abeparvovec-xioi); AND • Patient will not use in combination with other agents for SMA (e.g., onasemnogene abeparvovec, risdiplam, etc.); AND • Patient must not have advanced disease (complete limb paralysis, permanent ventilation support, etc.); AND Moda Health Plan, Inc. Medical Necessity Criteria Page 1/5 Proprietary & Confidential © 2021 Magellan Health, Inc. • Patient must have the following laboratory tests at baseline and prior to each administration*: platelet count, prothrombin time; activated -

2017 ANNUAL REPORT | Translating SCIENCE • Transforming LIVES OUR COMMITMENT Make Every Day Count at PTC, Patients Are at the Center of Everything We Do
20 YEARS OF COMMITMENT 2017 ANNUAL REPORT | Translating SCIENCE • Transforming LIVES OUR COMMITMENT Make every day count At PTC, patients are at the center of everything we do. We have the opportunity to support patients and families living with rare disorders through their journey. We know that every day matters and we are committed to making a difference. OUR SCIENCE Our scientists are finding new ways to regulate biology to control disease We have several scientific research platforms focused on modulating protein expression within the cell that we believe have the potential to address many rare genetic disorders. OUR PEOPLE Care for each other, our community, and for the needs of our patients At PTC, we are looking at drug discovery and development in a whole new light, bringing new technologies and approaches to developing medicines for patients living with rare disorders and cancer. We strive every day to be better than we were the day before. At PTC Therapeutics, it is our mission to provide access to best-in-class treatments for patients who have an unmet need. We are a science-led, global biopharmaceutical company focused on the discovery, development and commercialization of clinically-differentiated medicines that provide benefits to patients with rare disorders. Founded 20 years ago, PTC Therapeutics has successfully launched two rare disorder products and has a global commercial footprint. This success is the foundation that drives investment in a robust pipeline of transformative medicines and our mission to provide access to best-in-class treatments for patients who have an unmet medical need. As we celebrate our 20th year of bringing innovative therapies to patients affected by rare disorders, we reflect on our unwavering commitment to our patients, our science and our employees. -

Spinraza® (Nusinersen)
UnitedHealthcare® Commercial Medical Benefit Drug Policy Spinraza® (Nusinersen) Policy Number: 2021D0059I Effective Date: July 1, 2021 Instructions for Use Table of Contents Page Community Plan Policy Coverage Rationale ....................................................................... 1 • Spinraza® (Nusinersen) Documentation Requirements ...................................................... 3 Applicable Codes .......................................................................... 3 Background.................................................................................... 4 Benefit Considerations .................................................................. 5 Clinical Evidence ........................................................................... 5 U.S. Food and Drug Administration ............................................. 8 References ..................................................................................... 9 Policy History/Revision Information ........................................... 10 Instructions for Use ..................................................................... 11 Coverage Rationale See Benefit Considerations Spinraza® (nusinersen) is proven and medically necessary for the treatment of spinal muscular atrophy (SMA) in patients who meet all of the following criteria: 1-4,22,23 Initial Therapy Diagnosis of spinal muscular atrophy by, or in consultation with, a neurologist with expertise in the diagnosis of SMA; and Submission of medical records (e.g., chart notes, laboratory values) -

DRUGS REQUIRING PRIOR AUTHORIZATION in the MEDICAL BENEFIT Page 1
Effective Date: 08/01/2021 DRUGS REQUIRING PRIOR AUTHORIZATION IN THE MEDICAL BENEFIT Page 1 Therapeutic Category Drug Class Trade Name Generic Name HCPCS Procedure Code HCPCS Procedure Code Description Anti-infectives Antiretrovirals, HIV CABENUVA cabotegravir-rilpivirine C9077 Injection, cabotegravir and rilpivirine, 2mg/3mg Antithrombotic Agents von Willebrand Factor-Directed Antibody CABLIVI caplacizumab-yhdp C9047 Injection, caplacizumab-yhdp, 1 mg Cardiology Antilipemic EVKEEZA evinacumab-dgnb C9079 Injection, evinacumab-dgnb, 5 mg Cardiology Hemostatic Agent BERINERT c1 esterase J0597 Injection, C1 esterase inhibitor (human), Berinert, 10 units Cardiology Hemostatic Agent CINRYZE c1 esterase J0598 Injection, C1 esterase inhibitor (human), Cinryze, 10 units Cardiology Hemostatic Agent FIRAZYR icatibant J1744 Injection, icatibant, 1 mg Cardiology Hemostatic Agent HAEGARDA c1 esterase J0599 Injection, C1 esterase inhibitor (human), (Haegarda), 10 units Cardiology Hemostatic Agent ICATIBANT (generic) icatibant J1744 Injection, icatibant, 1 mg Cardiology Hemostatic Agent KALBITOR ecallantide J1290 Injection, ecallantide, 1 mg Cardiology Hemostatic Agent RUCONEST c1 esterase J0596 Injection, C1 esterase inhibitor (recombinant), Ruconest, 10 units Injection, lanadelumab-flyo, 1 mg (code may be used for Medicare when drug administered under Cardiology Hemostatic Agent TAKHZYRO lanadelumab-flyo J0593 direct supervision of a physician, not for use when drug is self-administered) Cardiology Pulmonary Arterial Hypertension EPOPROSTENOL (generic) -

New Brunswick Drug Plans Formulary
New Brunswick Drug Plans Formulary August 2019 Administered by Medavie Blue Cross on Behalf of the Government of New Brunswick TABLE OF CONTENTS Page Introduction.............................................................................................................................................I New Brunswick Drug Plans....................................................................................................................II Exclusions............................................................................................................................................IV Legend..................................................................................................................................................V Anatomical Therapeutic Chemical (ATC) Classification of Drugs A Alimentary Tract and Metabolism 1 B Blood and Blood Forming Organs 23 C Cardiovascular System 31 D Dermatologicals 81 G Genito Urinary System and Sex Hormones 89 H Systemic Hormonal Preparations excluding Sex Hormones 100 J Antiinfectives for Systemic Use 107 L Antineoplastic and Immunomodulating Agents 129 M Musculo-Skeletal System 147 N Nervous System 156 P Antiparasitic Products, Insecticides and Repellants 223 R Respiratory System 225 S Sensory Organs 234 V Various 240 Appendices I-A Abbreviations of Dosage forms.....................................................................A - 1 I-B Abbreviations of Routes................................................................................A - 4 I-C Abbreviations of Units...................................................................................A -

Prescription Drugs Requiring Prior Authorization
PRESCRIPTION DRUGS REQUIRING PRIOR AUTHORIZATION Revised 10/16 As part of our drug utilization management program, members must request and receive prior authorization for certain prescription drugs in order to use their prescription drug benefits. Below is a list of drugs that currently require prior authorization. This list will be updated periodically as new drugs that require prior authorization are introduced. As benefits may vary by group and individual plans, the inclusion of a medication on this list does not imply prescription drug coverage. The Schedule of Benefits contains a list of drug categories that require prior authorization. Prior authorization requests are processed by our pharmacy benefit manager, Express Scripts, Inc. (ESI). Physicians must call ESI to obtain an authorization. (1-800-842-2015). Drug Name Generic Name Drug Classification Abstral fentanyl citrate oral tablet Controlled Dangerous Substance Accu-Chek Test Strips blood glucose test strips Blood Glucose Test Strips Actemra tocilizumab Monoclonal Antibody Acthar corticotropin Hormone Actimmune interferon gamma 1b Interferon Actiq fentanyl citrate OTFC Controlled Dangerous Substance Adcirca tadalafil Pulmonary Vasodilator Adempas riociguat Pulmonary Vasodilator Adlyxin lixisenatide Type 2 Diabetes Advocate Test Strips blood glucose test strips Blood Glucose Test Strips Aerospan** flunisolide Corticosteroids (Inhaled) Afrezza insulin Insulin (inhaled) Ampyra dalfampridine Multiple Sclerosis Agent Altoprev** lovastatin Cholesterol Alvesco** ciclesonide Corticosteroids -

Circulating Biomarkers in Neuromuscular Disorders: What Is Known, What Is New
biomolecules Review Circulating Biomarkers in Neuromuscular Disorders: What Is Known, What Is New Andrea Barp 1,* , Amanda Ferrero 1, Silvia Casagrande 1,2 , Roberta Morini 1 and Riccardo Zuccarino 1 1 NeuroMuscular Omnicentre (NeMO) Trento, Villa Rosa Hospital, Via Spolverine 84, 38057 Pergine Valsugana, Italy; [email protected] (A.F.); [email protected] (S.C.); [email protected] (R.M.); [email protected] (R.Z.) 2 Department of Neurosciences, Drug and Child Health, University of Florence, Largo Brambilla 3, 50134 Florence, Italy * Correspondence: [email protected] Abstract: The urgent need for new therapies for some devastating neuromuscular diseases (NMDs), such as Duchenne muscular dystrophy or amyotrophic lateral sclerosis, has led to an intense search for new potential biomarkers. Biomarkers can be classified based on their clinical value into different categories: diagnostic biomarkers confirm the presence of a specific disease, prognostic biomarkers provide information about disease course, and therapeutic biomarkers are designed to predict or measure treatment response. Circulating biomarkers, as opposed to instrumental/invasive ones (e.g., muscle MRI or nerve ultrasound, muscle or nerve biopsy), are generally easier to access and less “time-consuming”. In addition to well-known creatine kinase, other promising molecules seem to be candidate biomarkers to improve the diagnosis, prognosis and prediction of therapeutic response, such as antibodies, neurofilaments, and microRNAs. However, there are some criticalities that can complicate their application: variability during the day, stability, and reliable performance metrics Citation: Barp, A.; Ferrero, A.; (e.g., accuracy, precision and reproducibility) across laboratories. In the present review, we discuss Casagrande, S.; Morini, R.; Zuccarino, the application of biochemical biomarkers (both validated and emerging) in the most common NMDs R. -

The Use of Ataluren in the Effective Management of Duchenne Muscular Dystrophy
Review Neuromuscular Diseases Early Diagnosis and Treatment – The Use of Ataluren in the Effective Management of Duchenne Muscular Dystrophy Eugenio Mercuri,1 Ros Quinlivan2 and Sylvie Tuffery-Giraud3 1. Catholic University, Rome, Italy; 2. Great Ormond Street Hospital and National Hospital for Neurology and Neurosurgery, London, UK; 3. Laboratory of Genetics of Rare Diseases (LGMR), University of Montpellier, Montpellier, France DOI: https://doi.org/10.17925/ENR.2018.13.1.31 he understanding of the natural history of Duchenne muscular dystrophy (DMD) is increasing rapidly and new treatments are emerging that have the potential to substantially improve the prognosis for patients with this disabling and life-shortening disease. For many, Thowever, there is a long delay between the appearance of symptoms and DMD diagnosis, which reduces the possibility of successful treatment. DMD results from mutations in the large dystrophin gene of which one-third are de novo mutations and two-thirds are inherited from a female carrier. Roughly 75% of mutations are large rearrangements and 25% are point mutations. Certain deletions and nonsense mutations can be treated whereas many other mutations cannot currently be treated. This emphasises the need for early genetic testing to identify the mutation, guide treatment and inform genetic counselling. Treatments for DMD include corticosteroids and more recently, ataluren has been approved in Europe, the first disease-modifying therapy for treating DMD caused by nonsense mutations. The use of ataluren in DMD is supported by positive results from phase IIb and phase III studies in which the treatment produced marked improvements in the 6-minute walk test, timed function tests such as the 10 m walk/run test and the 4-stair ascent/descent test compared with placebo. -

Animal Models of Duchenne Muscular Dystrophy: from Basic Mechanisms to Gene Therapy Joe W
© 2015. Published by The Company of Biologists Ltd | Disease Models & Mechanisms (2015) 8, 195-213 doi:10.1242/dmm.018424 REVIEW Animal models of Duchenne muscular dystrophy: from basic mechanisms to gene therapy Joe W. McGreevy1, Chady H. Hakim1, Mark A. McIntosh1 and Dongsheng Duan1,2,* ABSTRACT The identification of the disease-causing gene and the molecular Duchenne muscular dystrophy (DMD) is a progressive muscle- basis for the DMD and BMD phenotypes establishes the foundation wasting disorder. It is caused by loss-of-function mutations in the for DMD gene therapy (Fig. 2A). To mitigate muscle disease, one dystrophin gene. Currently, there is no cure. A highly promising can either restore the full-length transcript or express a truncated but therapeutic strategy is to replace or repair the defective dystrophin in-frame dystrophin gene (Duan, 2011; Goyenvalle et al., 2011; gene by gene therapy. Numerous animal models of DMD have been Konieczny et al., 2013; Mendell et al., 2012; Verhaart and Aartsma- developed over the last 30 years, ranging from invertebrate to large Rus, 2012). Several gene therapy strategies are currently under mammalian models. mdx mice are the most commonly employed development. They include replacing the mutated gene with a models in DMD research and have been used to lay the groundwork functional candidate gene (gene replacement) or repairing the for DMD gene therapy. After ~30 years of development, the field has defective gene by targeted correction and exon skipping (gene reached the stage at which the results in mdx mice can be validated repair). Currently, adeno-associated virus (AAV)-mediated gene and scaled-up in symptomatic large animals. -
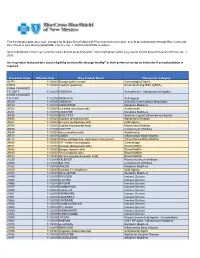
Procedure Code Effective Date Drug Product Name* Therapeutic
This list includes procedure code changes for Medical Benefit Specialty Pharmacy that may require benefit preauthorization through Blue Cross and Blue Shield of New Mexico (BCBSNM) effective Jan. 1, 2020 for BCBSNM members. Items highlighted in blue may currently require benefit preauthorization. Items highlighted yellow may require benefit preauthorization effective Jan. 1, 2020. It is imperative that providers check eligibility and benefits through Availity® or their preferred vendor to determine if preauthorization is required. Procedure Code Effective Date Drug Product Name* Therapeutic Category 90378 1/1/2020 Synagis (palivizumab) Immunological Agent C9036 1/1/2020 Onpattro (patisiran) Small interfering RNA (siRNA) C9466 CHANGED TO J0517 1/1/2019 FASENRA Antiasthmatic - Monoclonal Antibodies C9493 CHANGED TO J1301 1/1/2019 RADICAVA ALS Agents J0129 1/1/2019 ORENCIA Selective Costimulation Modulators J0180 1/1/2019 FABRAZYME Metabolic Modifiers J0202 1/1/2020 Lemtrada (alemtuzumab) Autoimmune J0221 1/1/2019 LUMIZYME Metabolic Modifiers J0490 1/1/2019 BENLYSTA Systemic Lupus Erythematosus Agents J0565 1/1/2020 Zinplava (bezlotoxumab) Monoclonal Antibody J0567 1/1/2020 Brineura (cerliponase alfa) CNS Agents J0584 1/1/2020 Crysvita (burosumab-twza) Monoclonal Antibody J0598 1/1/2019 CINRYZE Complement Inhibitors J0638 1/1/2020 Ilaris (canakinumab) Autoimmune J0717 1/1/2019 CIMZIA Inflammatory Bowel Agents J0775 1/1/2020 Xiaflex (collagenase, clostridium histolyticum) Tissue Permeability Modifier J0800 1/1/2020 H.P. Acthar (corticotropin) -

Corporate Medical Policy
Corporate Medical Policy Nusinersen (Spinraza™) File Name: nusinersen_spinraza Origination: 03/2017 Last CAP Review: 10/2020 Next CAP Review: 10/2021 Last Review: 10/2020 Description of Procedure or Service Spinal muscular atrophy (SMA) is an inherited disorder caused by homozygous deletions or variants in the SMN1 gene. As a consequence of low levels of SMN1 protein, the motor neurons in spinal cord degenerate, resulting in atrophy of the voluntary muscles of the limbs and trunk. Nusinersen is a synthetic antisense oligonucleotide designed to bind to a specific sequence in exon 7 of the SMN2 transcript causing the inclusion of exon 7 in the SMN2 transcript, leading to production of full length functional SMN2 protein, which demonstrates similarity to SMN1. SPINAL MUSCULAR ATROPHY Spinal muscular atrophy (SMA) is a rare autosomal recessive genetic disorder caused by homozygous deletions or variants in the SMN1 gene in chromosome 5. This gene is responsible for producing the “survival of motor neuron” protein (SMN1). As a consequence of absent or low levels of this protein, the motoneurons in the spinal cord degenerate, resulting in atrophy of the voluntary muscles of the limbs and trunk. During early development, these muscles are necessary for crawling, walking, sitting up, and head control. The more severe types of SMA can also affect muscles involved in feeding, swallowing, and breathing. The exact role of the SMN protein in motoneurons has not been completely elucidated and levels of the SMN protein required for optimal functioning are unknown. SMN2 is a nearly identical modifying gene capable of producing compensatory SMN protein. -

Outpatient Drug Services Handbook
Texas Medicaid Provider Procedures Manual December 2020 Provider Handbooks Outpatient Drug Services Handbook The Texas Medicaid & Healthcare Partnership (TMHP) is the claims administrator for Texas Medicaid under contract with the Texas Health and Human Services Commission. TEXAS MEDICAID PROVIDER PROCEDURES MANUAL: VOL. 2 DECEMBER 2020 OUTPATIENT DRUG SERVICES HANDBOOK Table of Contents 1 General Information . 7 1.1 About the Vendor Drug Program. 7 1.2 Pharmacy Enrollment . 8 1.3 Program Contact Information. 8 2 Enrollment . 8 3 Services, Benefits, Limitations, and Prior Authorization. .8 3.1 Prior Authorization Requests . 9 3.2 Electronic Signatures in Prior Authorizations . 9 4 Reimbursement. .10 5 Injectable Medications as a Pharmacy Benefit. .11 6 National Drug Code (NDC) . .12 6.1 Calculating Billable HCPCS and NDC Units . .12 6.1.1 Single-Dose Vials Calculation Examples . 12 6.1.2 Multi-Dose Vials Calculation Examples . 13 6.1.3 Single and Multi-Use Vials . 13 6.1.4 Nonspecific, Unlisted, or Miscellaneous Procedure Codes . 14 7 Outpatient Drugs—Benefits and Limitations. .15 7.1 Abatacept (Orencia) . .15 7.1.1 Prior Authorization for Abatacept (Orencia) . 15 7.2 Adalimumab. .16 7.3 Ado-trastuzumab entansine (Kadcyla). .17 7.4 Alglucosidase Alfa (Myozyme) . .18 7.5 Amifostine . .18 7.6 Antibiotics and Steroids . .22 7.7 Antisense Oligonucleotides (eteplirsen, golodirsen, and nusinersen) . .22 7.7.1 Prior Authorization Requirements. 22 7.7.1.1 Initial Requests (for all Antisense Oligonucleotides) . 23 7.7.1.2 Recertification/Extension Requests (for all Antisense Oligonucleotides). 25 7.7.1.3 Exclusions . 25 7.8 Aripiprazole Lauroxil, (Aristada Initio).