Biochemical and Structural Studies of Microbial
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Glycerol Dehydrogenase from Gluconobacter Industrius
Agric. Biol Chem., 49 (4), 1001 -1010, 1985 1001 Solubilization, Purification and Properties of Membrane-bound Glycerol Dehydrogenase from Gluconobacter industrius Minoru Ameyama,Emiko Shinagawa, Kazunobu Matsushita and Osao Adachi Laboratory of Applied Microbiology, Department of Agricultural Chemistry, Faculty of Agriculture, Yamaguchi University, Yamaguchi 753, Japan Received July 30, 1984 Membrane-bound glycerol dehydrogenase was solubilized and purified about 100-fold from the membraneof Gluconobacter industrius IFO 3260 grown on a glycerol-glutamate medium. Solubilization of the enzyme was successfully achieved by use of 0.5% dimethyldodecylamineoxide in 0.05 m Tris-HCl, pH 8.0. Alcohol dehydrogenase and D-glucose dehydrogenase, which were abundantly formed in the same bacterial membrane, were eliminated on solubilization. Glycerol dehydrogenase was further purified through fractionation with polyethylene glycol 6000. The enzymeshowed a broad substrate specificity and various kinds of polyhydroxyl alcohols, in addition to glycerol, were rapidly oxidized in the presence of 2,6-dichlorophenolindophenoi and phenazine methosulfate as the electron acceptor but NADand NADPwere inert. The enzyme was proved to be a quinoprotein in which pyrroloquinoline quinone functioned as the prosthetic group. The first report on microbial oxidation of localization of the oxidase system in cells of G. glycerol to dihydroxyacetone was by Bertrand liquefaciens and found that the oxidation of with a strain capable of L-sorbose fermen- glycerol and raeso-erythritol -

Toxicological Profile for Acetone Draft for Public Comment
ACETONE 1 Toxicological Profile for Acetone Draft for Public Comment July 2021 ***DRAFT FOR PUBLIC COMMENT*** ACETONE ii DISCLAIMER Use of trade names is for identification only and does not imply endorsement by the Agency for Toxic Substances and Disease Registry, the Public Health Service, or the U.S. Department of Health and Human Services. This information is distributed solely for the purpose of pre dissemination public comment under applicable information quality guidelines. It has not been formally disseminated by the Agency for Toxic Substances and Disease Registry. It does not represent and should not be construed to represent any agency determination or policy. ***DRAFT FOR PUBLIC COMMENT*** ACETONE iii FOREWORD This toxicological profile is prepared in accordance with guidelines developed by the Agency for Toxic Substances and Disease Registry (ATSDR) and the Environmental Protection Agency (EPA). The original guidelines were published in the Federal Register on April 17, 1987. Each profile will be revised and republished as necessary. The ATSDR toxicological profile succinctly characterizes the toxicologic and adverse health effects information for these toxic substances described therein. Each peer-reviewed profile identifies and reviews the key literature that describes a substance's toxicologic properties. Other pertinent literature is also presented, but is described in less detail than the key studies. The profile is not intended to be an exhaustive document; however, more comprehensive sources of specialty information are referenced. The focus of the profiles is on health and toxicologic information; therefore, each toxicological profile begins with a relevance to public health discussion which would allow a public health professional to make a real-time determination of whether the presence of a particular substance in the environment poses a potential threat to human health. -

High-Yield Anaerobic Succinate Production by Strategically
Meng et al. Microb Cell Fact (2016) 15:141 DOI 10.1186/s12934-016-0536-1 Microbial Cell Factories RESEARCH Open Access High‑yield anaerobic succinate production by strategically regulating multiple metabolic pathways based on stoichiometric maximum in Escherichia coli Jiao Meng1,2,3†, Baiyun Wang1,2,3†, Dingyu Liu1,2,3, Tao Chen1,2,3,4, Zhiwen Wang1,2,3* and Xueming Zhao1,2,3 Abstract Background: Succinate has been identified by the U.S. Department of Energy as one of the top 12 building block chemicals, which can be used as a specialty chemical in the agricultural, food, and pharmaceutical industries. Escheri- chia coli are now one of the most important succinate producing candidates. However, the stoichiometric maximum succinate yield under anaerobic conditions through the reductive branch of the TCA cycle is restricted by NADH sup- ply in E. coli. Results: In the present work, we report a rational approach to increase succinate yield by regulating NADH supply via pentose phosphate (PP) pathway and enhancing flux towards succinate. The deregulated genes zwf243 (encod- ing glucose-6-phosphate dehydrogenase) and gnd361 (encoding 6-phosphogluconate dehydrogenase) involved in NADPH generation from Corynebacterium glutamicum were firstly introduced into E. coli for succinate production. Co-expression of beneficial mutated dehydrogenases, which removed feedback inhibition in the oxidative part of the PP pathway, increased succinate yield from 1.01 to 1.16 mol/mol glucose. Three critical genes, pgl (encoding 6-phos- phogluconolactonase), tktA (encoding transketolase) and talB (encoding transaldolase) were then overexpressed to redirect more carbon flux towards PP pathway and further improved succinate yield to 1.21 mol/mol glucose. -

The Diversity of Microbial Aldo/Keto Reductases from Escherichia Coli K12
Lapthorn, A. J., Zhu, X., and Ellis, E. M. (2013) The diversity of microbial aldo/keto reductases from Escherichia coli K12. Chemico-Biological Interactions, 202(1-3), pp. 168-177. (doi:10.1016/j.cbi.2012.10.008) There may be differences between this version and the published version. You are advised to consult the publisher’s version if you wish to cite from it. http://eprints.gla.ac.uk/124204/ Deposited on: 07 October 2016 Enlighten – Research publications by members of the University of Glasgow http://eprints.gla.ac.uk The diversity of microbial aldo/keto reductases from Escherichia coli K12 Adrian J. Lapthorn 1, Xiaofeng Zhu 2,3 and Elizabeth M. Ellis 3 1 School of Chemistry, Joseph Black Building, University of Glasgow, Glasgow G12 8QQ 2 College of Life Science and State Key Laboratory of Biotherapy and Cancer Centre, Sichuan University, Chengdu, China 3 Strathclyde Institute of Pharmacy and Biomedical Sciences, 161 Cathedral Street, Glasgow, G4 0RE Corresponding author: Adrian J. Lapthorn School of Chemistry, Joseph Black Building, University of Glasgow, Glasgow G12 8QQ Tel: +44 141-330 5940 Fax: +44 141-330 4888 E-mail: [email protected] Key Words: Aldo-Keto reductases, methylglyoxal reductase, 2,5-diketo-D-gluconate reductase, tyrosine auxotrophy suppressor protein, L-glyceraldehyde 3-phosphate reductase, AKR quaternary structure. Abstract The genome of Escherichia coli K12 contains 9 open reading frames encoding aldo/keto reductases (AKR) that are differentially regulated and sequence diverse. A significant amount of data is available for the E. coli AKRs through the availability of gene knockouts and gene expression studies, which adds to the biochemical and kinetic data. -

Engineering of Glycerol Utilization in Gluconobacter Oxydans 621H For
Yan et al. Microb Cell Fact (2018) 17:158 https://doi.org/10.1186/s12934-018-1001-0 Microbial Cell Factories RESEARCH Open Access Engineering of glycerol utilization in Gluconobacter oxydans 621H for biocatalyst preparation in a low‑cost way Jinxin Yan1, Jing Xu1,3, Menghao Cao1, Zhong Li1, Chengpeng Xu1, Xinyu Wang1, Chunyu Yang1, Ping Xu2, Chao Gao1 and Cuiqing Ma1* Abstract Background: Whole cells of Gluconobacter oxydans are widely used in various biocatalytic processes. Sorbitol at high concentrations is commonly used in complex media to prepare biocatalysts. Exploiting an alternative process for preparation of biocatalysts with low cost substrates is of importance for industrial applications. Results: G. oxydans 621H was confrmed to have the ability to grow in mineral salts medium with glycerol, an inevitable waste generated from industry of biofuels, as the sole carbon source. Based on the glycerol utilization mechanism elucidated in this study, the major polyol dehydrogenase (GOX0854) and the membrane-bound alcohol dehydrogenase (GOX1068) can competitively utilize glycerol but play no obvious roles in the biocatalyst prepara- tion. Thus, the genes related to these two enzymes were deleted. Whole cells of G. oxydans ∆GOX1068∆GOX0854 can be prepared from glycerol with a 2.4-fold higher biomass yield than that of G. oxydans 621H. Using whole cells of G. 1 1 oxydans ∆GOX1068∆GOX0854 as the biocatalyst, 61.6 g L− xylonate was produced from 58.4 g L− xylose at a yield of 1 1.05 g g− . Conclusion: This process is an example of efcient preparation of whole cells of G. oxydans with reduced cost. -

Characterization of Glycerol Dehydrogenase from Thermoanaerobacterium Thermosaccharolyticum DSM 571 and GGG Motif Identification
J. Microbiol. Biotechnol. (2016), 26(6), 1077–1086 http://dx.doi.org/10.4014/jmb.1512.12051 Research Article Review jmb Characterization of Glycerol Dehydrogenase from Thermoanaerobacterium thermosaccharolyticum DSM 571 and GGG Motif Identification Liangliang Wang1,2, Jiajun Wang1,2, Hao Shi1,2, Huaxiang Gu1,2, Yu Zhang1,2, Xun Li1,2, and Fei Wang1,2* 1College of Chemical Engineering, Nanjing Forestry University, Nanjing 210037, P.R. China 2Jiangsu Key Laboratory of Biomass-Based Green Fuels and Chemicals, Nanjing 210037, P.R. China Received: December 17, 2015 Revised: March 5, 2016 Glycerol dehydrogenases (GlyDHs) are essential for glycerol metabolism in vivo, catalyzing Accepted: March 9, 2016 its reversible reduction to 1,3-dihydroxypropranone (DHA). The gldA gene encoding a putative GlyDH was cloned from Thermoanaerobacterium thermosaccharolyticum DSM 571 (TtGlyDH) and expressed in Escherichia coli. The presence of Mn2+ enhanced its enzymatic 171 254 271 First published online activity by 79.5%. Three highly conserved residues (Asp , His , and His ) in TtGlyDH were March 14, 2016 associated with metal ion binding. Based on an investigation of glycerol oxidation and DHA *Corresponding author reduction, TtGlyDH showed maximum activity towards glycerol at 60°C and pH 8.0 and Phone: +86-25-85427649; towards DHA at 60°C and pH 6.0. DHA reduction was the dominant reaction, with a lower Fax: +86-25-85427649; K of 1.08 ± 0.13 mM and V of 0.0053 ± 0.0001 mM/s, compared with glycerol oxidation, E-mail: [email protected] m(DHA) max with a Km(glycerol) of 30.29 ± 3.42 mM and Vmax of 0.042 ± 0.002 mM/s. -

Molecular Properties of Membrane-Bound FAD-Containing D-Sorbitol Dehydrogenase from Thermotolerant Gluconobacter Frateurii Isolated from Thailand
Biosci. Biotechnol. Biochem., 69 (6), 1120–1129, 2005 Molecular Properties of Membrane-Bound FAD-Containing D-Sorbitol Dehydrogenase from Thermotolerant Gluconobacter frateurii Isolated from Thailand y Hirohide TOYAMA,1; Wichai SOEMPHOL,1 Duangtip MOONMANGMEE,2 Osao ADACHI,1 and Kazunobu MATSUSHITA1 1Department of Biological Chemistry, Faculty of Agriculture, Yamaguchi University, Yamaguchi 753-8515, Japan 2Department of Microbiology, Faculty of Science, King Mongkut’s University of Technology Thonburi, Prachauthit Road., Tungkru, Bangkok 10140, Thailand Received December 24, 2004; Accepted March 14, 2005 There are two types of membrane-bound D-sorbitol ism leads to applications in industry for fermentation of dehydrogenase (SLDH) reported: PQQ–SLDH, having valuable products such as L-sorbose, dihydroxyacetone, pyrroloquinoline quinone (PQQ), and FAD–SLDH, D-gluconate, and keto-D-gluconates.3,4) containing FAD and heme c as the prosthetic groups. L-Sorbose is an important intermediate in the indus- FAD–SLDH was purified and characterized from the trial production of vitamin C. The membrane-bound PQQ–SLDH mutant strain of a thermotolerant Gluco- D-sorbitol dehydrogenase (SLDH) has been considered nobacter frateurii, having molecular mass of 61.5 kDa, to play a main role in L-sorbose fermentation.5) In 52 kDa, and 22 kDa. The enzyme properties were quite Gluconobacter strains, two types of membrane-bound similar to those of the enzyme from mesophilic G. oxy- D-sorbitol dehydrogenases have been purified and well dans IFO 3254. This enzyme was shown to be inducible characterized. One was purified from G. suboxydans var by D-sorbitol, but not PQQ–SLDH. The oxidation IFO 32546) as flavohemoprotein, containing a cova- product of FAD–SLDH from D-sorbitol was identified lently bound FAD having three subunits with molecular as L-sorbose. -

Transcriptional Regulation Mechanisms Involved in Azole Resistance in Candida Species: Focusing on the Transcription Factors Rpn4 and Mrr1
Transcriptional regulation mechanisms involved in azole resistance in Candida species: focusing on the transcription factors Rpn4 and Mrr1 Raquel da Silva Califórnia Thesis to obtain the Master of Science Degree in Biotechnology Supervisor: Prof. Dr. Miguel Nobre Parreira Cacho Teixeira Examination Committee Chairperson: Prof. Dr. Ana Cristina Anjinho Madeira Viegas Supervisor: Prof. Dr. Miguel Nobre Parreira Cacho Teixeira Member of the Committee: Dr. Catarina Isabel Ribeiro Pimentel October 2018 ii Acknowledgements For me the development of this thesis was very challenging and involved a very extensive work, whose purpose would not have been reached without the help of some people I will mention below. First of all, I would like to thank my supervisor Professor Miguel Teixeira for the opportunity given by accepting me in his team and in this project. His tremendous support, guidance and motivation, always available to help, were crucial for the success of this work. I would like to thank Professor Isabel Sá-Correia for giving me the chance to join the Biological Sciences Research Group to develop my master thesis work. The achievement of this thesis required an indispensable help from several parts, which deserve my recognition. For the collaboration in the transcriptomic analysis herein accomplished, I thank Professor Geraldine Butler and her team, from University College of Dublin. For the supply of Candida glabrata mutants used in this work, I have to thank Professor Hiroji Chibana, from University of Chiba, Japan. For the study developed in HPLC analysis of ergosterol levels, I thank also Professor Nuno Mira for his availability and assistance. My gratitude should also be expressed towards my colleague, Pedro Pais, for the great help he has given me throughout this period, always available to help and explain anything. -
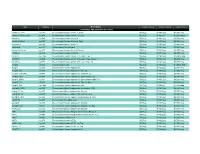
Prospec (Product
Name Catalog # Description Customer Price-A Customer Price-B Customer Price-C CYTOKINES AND GROWTH FACTORS Activin A, Active cyt-145 Recombinant Human Activin-A, Active $50/2µg $130/10µg $3,500/1mg Activin A, Plant-Active cyt-414 Recombinant Human Activin-A Active $50/1µg $130/5µg $1,500/100µg Activin A cyt-569 Recombinant Human Activin-A $50/2µg $130/10µg $2,700/1mg Activin A, Plant cyt-052 Recombinant Human Activin-A, Plant $50/2µg $130/10µg $4,800/1mg mActivin A cyt-146 Recombinant Mouse Activin-A $50/2µg $130/10µg $3,500/1mg rActivin A cyt-147 Recombinant Rat Activin-A $50/2µg $130/10µg $3,500/1mg Activin B, Active cyt-057 Recombinant Human Activin-B Active $50/2µg $130/10µg $5,200/1mg Activin B cyt-058 Recombinant Human Activin-B $50/2µg $130/10µg $4,800/1mg ACVR1 cyt-1140 Recombinant Human Activin A Receptor Type 1 $50/2µg $130/10µg $1,000/0.1mg ACVRL1 cyt-920 Recombinant Human Activin A Receptor Type II-Like 1 $50/2µg $130/10µg $5,200/1mg ACVR2A cyt-976 Recombinant Human Activin A Receptor Type 2A $50/2µg $130/10µg $5,200/1mg Acrp30 cyt-024 Human Adiponectin $50/2µg $130/10µg $1,000/0.1mg Acrp30 cyt-280 Recombinant Human Adiponectin $50/5µg $130/25µg $2,700/1mg Acrp30, His cyt-433 Recombinant Human Adiponectin, His tag $50/10µg $130/50µg $1,900/1mg Acrp30 (108-244) cyt-073 Recombinant Human Adiponectin (108-244 a.a.) $50/5µg $130/25µg $2,700/1mg Acrp30, HEK cyt-434 Recombinant Human Adiponectin glycosilated, HEK $50/2µg $130/10µg $4,680/1mg Acrp30, HMW cyt-764 Recombinant Human Adiponectin glycosilated, HMW Rich $50/2µg $130/10µg -

Prospec Product Catalog 2019
ProSpec Product Catalog 2019 QUALITY PROTEINS for scientific discoveries WE'VE GOT CHEMISTRY BRIGHTEN YOUR RESEARCH part for life sciences 6,000 proteins at your fingertips List by Molecule Name List by Molecule Name 제품별 자세한 설명은 www.prospecbio.com을 방문하세요. 부가세 별도 Molecule 제품번호 제품명 Size A / 가격(원) Size B / 가격(원) Size C / 가격(원) Name 1F8 Chagas cch-002 Recombinant 1F8 Chagas 5µg / 90,500 20µg / 199,000 1mg / 5,110,500 4-1BBL cyt-649 Recombinant Human 4-1BB Ligand, His Tag 5µg / 112,200 20µg / 220,800 1mg / 3,793,200 4-1BBL cyt-149 Recombinant Human 4-1BB Ligand 5µg / 90,500 20µg / 199,000 1mg / 3,771,400 4-1BBR cyt-916 Recombinant Mouse 4-1BB Receptor 1µg / 112,200 5µg / 220,800 50µg / 1,701,000 4-1BBR cyt-463 Recombinant Human 4-1BB Receptor 5µg / 90,500 20µg / 199,000 1mg / 3,771,400 4-1BBR cyt-137 Recombinant Human 4-1BB Receptor His Tag 5µg / 112,200 20µg / 220,800 1mg / 5,016,800 4-1BBR cyt-931 Recombinant Human 4-1BB Receptor sf9 2µg / 112,200 10µg / 220,800 1mg / 7,227,300 A2LD1 pro-172 Recombinant Human AIG2-Like Domain 1 2µg / 112,200 10µg / 220,800 1mg / 7,227,300 A2M pro-551 Human Alpha-2 Macroglobulin Protein 200µg / 90,500 1mg / 317,500 10mg / 2,389,800 Recombinant Human Alpha & Gamma-Adaptin Binding AAGAB pro-1479 5µg / 112,200 20µg / 220,800 1mg / 3,793,200 Protein Recombinant Human Adipogenesis Associated, Mth938 AAMDC pro-2116 5µg / 112,200 20µg / 220,800 1mg / 3,793,200 Domain Containing AARS enz-305 Recombinant Human Alanyl t-RNA Synthetase 5µg / 112,200 20µg / 220,800 1mg / 5,372,000 Recombinant Human Aminoadipate-Semialdehyde -

Application of High-Resolution Mass Spectrometry and a Theoretical
pubs.acs.org/est Article Application of High-Resolution Mass Spectrometry and a Theoretical Model to the Quantification of Multifunctional Carbonyls and Organic Acids in e‑Cigarette Aerosol Yichen Li, Amanda E. Burns, Guy J.P. Burke, Morgan E. Poindexter, Amy K. Madl, Kent E. Pinkerton, and Tran B. Nguyen* Cite This: Environ. Sci. Technol. 2020, 54, 5640−5650 Read Online ACCESS Metrics & More Article Recommendations *sı Supporting Information ABSTRACT: Electronic (e-) cigarette aerosol (particle and gas) is a complex mixture of chemicals, of which the profile is highly dependent on device operating parameters and e-liquid flavor formulation. The thermal degradation of the e-liquid solvents propylene glycol and glycerol often generates multifunctional carbonyls that are challenging to quantify because of unavailability of standards. We developed a theoretical method to calculate the relative electrospray ionization sensitivities of hydrazones of organic acids and carbonyls with 2,4-dinitrophe- Δ nylhydrazine based on their gas-phase basicities ( Gdeprotonation). This method enabled quantification by high-performance liquid chromatography−high- resolution mass spectrometry HPLC-HRMS in the absence of chemical standards. Accurate mass and tandem multistage MS (MSn) were used for structure identification of vaping products. We quantified five simple carbonyls, six hydroxycarbonyls, four dicarbonyls, three acids, and one phenolic carbonyl in the e-cigarette aerosol with Classic Tobacco flavor. Our results suggest that hydroxycarbonyls, such as hydroxyacetone, lactaldehyde, and dihydroxyacetone can be significant components in e-cigarette aerosols but have received less attention in the literature and have poorly understood health effects. The data support the radical-mediated e-liquid thermal degradation scheme that has been previously proposed and emphasize the need for more research on the chemistry and toxicology of the complex product formation in e-cigarette aerosols. -

Kinetic Analysis of the Dimerization and Disproportionation of Aqueous Glyoxal Alfred Richard Fratzke Jr
Iowa State University Capstones, Theses and Retrospective Theses and Dissertations Dissertations 1985 Kinetic analysis of the dimerization and disproportionation of aqueous glyoxal Alfred Richard Fratzke Jr. Iowa State University Follow this and additional works at: https://lib.dr.iastate.edu/rtd Part of the Chemical Engineering Commons Recommended Citation Fratzke, Alfred Richard Jr., "Kinetic analysis of the dimerization and disproportionation of aqueous glyoxal" (1985). Retrospective Theses and Dissertations. 7845. https://lib.dr.iastate.edu/rtd/7845 This Dissertation is brought to you for free and open access by the Iowa State University Capstones, Theses and Dissertations at Iowa State University Digital Repository. It has been accepted for inclusion in Retrospective Theses and Dissertations by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. INFORMATION TO USERS This reproduction was made from a copy of a document sent to us for microfilming. While the most advanced technology has been used to photograph and reproduce this document, the quality of the reproduction is heavily dependent upon the quality of the material submitted. The following explanation of techniques is provided to help clarify markings or notations which may appear on this reproduction. 1.The sign or "target" for pages apparently lacking from the document photographed is "Missing Page(s)". If it was possible to obtain the missing page(s) or section, they are spliced into the film along with adjacent pages. This may have necessitated cutting through an image and duplicating adjacent pages to assure complete continuity. 2. When an image on the film is obliterated with a round black mark, it is an indication of either blurred copy because of movement during exposure, duplicate copy, or copyrighted materials that should not have been filmed.