Structural Intermediates and Directionality of the Swiveling
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Phosphotransferase Activity of Liver Mitochondria (Oxidative Phosphorylation/Adenine Nucleotide Ni-Oxides/Substrate Specificity/In Vivo Phosphorylation) G
Proc. Nat. Acad. Sci. USA Vol. 71, No. 11, pp. 4630-4634, November 1974 Participation of N1-Oxide Derivatives of Adenine Nucleotides in the Phosphotransferase Activity of Liver Mitochondria (oxidative phosphorylation/adenine nucleotide Ni-oxides/substrate specificity/in vivo phosphorylation) G. JEBELEANU, N. G. TY, H. H. MANTSCH*, 0. BARZU, G. NIACt, AND I. ABRUDAN Department of Biochemistry, Medical and Pharmaceutical Institute, Cluj, * Institute of Chemistry, University of Cluj, Cluj, and t Department of Physical Chemistry, University of Craiova, Craiova, Romania Communicated by Henry Lardy, September 3, 1974 ABSTRACT The modified adenine nucleotides ATP- rivatives of adenine nucleotides once produced become "ac- NO, ADP-NO, and AMP-NO were tested as potential tive" components of the mitochondrial or cellular adenylate substrates and/or inhibitors of mitochondrial phospho- transferases. ADP-NO is not recognized by the translocase pool. membrane; system located in the inner mitochondrial AND METHODS however, it is rapidly phosphorylated to ATP-NO in the MATERIALS outer compartment of mitochondria, by way of the The following commercially available chemicals were used: nucleosidediplhosphate kinase (EC 2.7.4.6) reaction, pro-' de- vitded there is sufficient ATP in the mitochondria. AMP- crystalline bovine serum albumin, glucose-6-phosphate NO is not phosphorylated by liver mitochondria to the hydrogenase (Glc-6-P dehydrogenase; EC 1.1.1.49; BDH corresponding nucleoside diphosphate; it cannot serve as Chemicals, Ltd.), yeast hexokinase (EC 2.7.1.1) (Nutritional substrate for adenylate kinase (EC 2.7.4.3). ATP-NO and Biochemicals, Cleveland), (log muscle lactate dehydrogenase ADP-NO, however, are substrates of this enzyme. -

Pyruvate Kinase: Function, Regulation and Role in Cancer
Pyruvate kinase: Function, regulation and role in cancer The MIT Faculty has made this article openly available. Please share how this access benefits you. Your story matters. Citation Israelsen, William J., and Matthew G. Vander Heiden. “Pyruvate Kinase: Function, Regulation and Role in Cancer.” Seminars in Cell & Developmental Biology 43 (2015): 43–51. As Published http://dx.doi.org/10.1016/j.semcdb.2015.08.004 Publisher Elsevier Version Author's final manuscript Citable link http://hdl.handle.net/1721.1/105833 Terms of Use Creative Commons Attribution-NonCommercial-NoDerivs License Detailed Terms http://creativecommons.org/licenses/by-nc-nd/4.0/ HHS Public Access Author manuscript Author Manuscript Author ManuscriptSemin Cell Author Manuscript Dev Biol. Author Author Manuscript manuscript; available in PMC 2016 August 13. Published in final edited form as: Semin Cell Dev Biol. 2015 July ; 43: 43–51. doi:10.1016/j.semcdb.2015.08.004. Pyruvate kinase: function, regulation and role in cancer William J. Israelsena,1,* and Matthew G. Vander Heidena,b,* aKoch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, Cambridge, MA 02139, USA bDepartment of Medical Oncology, Dana-Farber Cancer Institute, Boston, MA 02115, USA Abstract Pyruvate kinase is an enzyme that catalyzes the conversion of phosphoenolpyruvate and ADP to pyruvate and ATP in glycolysis and plays a role in regulating cell metabolism. There are four mammalian pyruvate kinase isoforms with unique tissue expression patterns and regulatory properties. The M2 isoform of pyruvate kinase (PKM2) supports anabolic metabolism and is expressed both in cancer and normal tissue. The enzymatic activity of PKM2 is allosterically regulated by both intracellular signaling pathways and metabolites; PKM2 thus integrates signaling and metabolic inputs to modulate glucose metabolism according to the needs of the cell. -

Part I Principles of Enzyme Catalysis
j1 Part I Principles of Enzyme Catalysis Enzyme Catalysis in Organic Synthesis, Third Edition. Edited by Karlheinz Drauz, Harald Groger,€ and Oliver May. Ó 2012 Wiley-VCH Verlag GmbH & Co. KGaA. Published 2012 by Wiley-VCH Verlag GmbH & Co. KGaA. j3 1 Introduction – Principles and Historical Landmarks of Enzyme Catalysis in Organic Synthesis Harald Gr€oger and Yasuhisa Asano 1.1 General Remarks Enzyme catalysis in organic synthesis – behind this term stands a technology that today is widely recognized as a first choice opportunity in the preparation of a wide range of chemical compounds. Notably, this is true not only for academic syntheses but also for industrial-scale applications [1]. For numerous molecules the synthetic routes based on enzyme catalysis have turned out to be competitive (and often superior!) compared with classic chemicalaswellaschemocatalyticsynthetic approaches. Thus, enzymatic catalysis is increasingly recognized by organic chemists in both academia and industry as an attractive synthetic tool besides the traditional organic disciplines such as classic synthesis, metal catalysis, and organocatalysis [2]. By means of enzymes a broad range of transformations relevant in organic chemistry can be catalyzed, including, for example, redox reactions, carbon–carbon bond forming reactions, and hydrolytic reactions. Nonetheless, for a long time enzyme catalysis was not realized as a first choice option in organic synthesis. Organic chemists did not use enzymes as catalysts for their envisioned syntheses because of observed (or assumed) disadvantages such as narrow substrate range, limited stability of enzymes under organic reaction conditions, low efficiency when using wild-type strains, and diluted substrate and product solutions, thus leading to non-satisfactory volumetric productivities. -
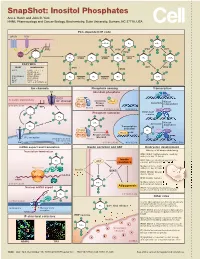
Snapshot: Inositol Phosphates Ace J
SnapShot: Inositol Phosphates Ace J. Hatch and John D. York HHMI, Pharmacology and Cancer Biology, Biochemistry, Duke University, Durham, NC 27710, USA PLC-dependent IP code GPCR RTK O O O O O O 5-PP-IP4 IP4 5-IP7 O O O O O O PIP2 O IP6K O IP6K O VIP1 O O O 2 O ITPK1 O 13 O PLC 2 O O O O O O O O 4 6 13 O 5 IP3 IPMK IP4 IPMK IP5 IPK1 IP6 1,5-IP8 4 6 O O 5 O O O O O O O O ENZYMES O O O O O O YEAST MAMMALIAN IP3K VIP1 IP6K IPMK PLC1 PLCβ, γ, δ, ε, ζ, η O - IP3KA, B, C - ITPK1 (IP56K) O O O O O O O O IPK2(ARG82) IPMK (IPK2) IP4 IP3 IP4 1-IP7 IPK1 IPK1 (IP5K) INPP5 ITPK1 KCS1 IP6K1, 2, 3 O O O O O O VIP1 VIP1, 2 (PPIP5K1, 2) O O Ion channels Phosphate sensing Transcription Cl- Abundant phosphate MCM1 ARG80 CIC3 P PLASMA MEMBRANE - Pho80 Cl channel Pho4 Kinase Kinase Assembly Pho85 independent CYTOPLASM activity 2 O PIP2 Pho81 13 CYTOPLASM NUCLEUS IPK2 ARG81 4 6 Phosphate starvation MCM1-ArgR O 5 O complex O O IP4 O O O O O O O 1-IP7 Kinase Activation dependent IP3 O O Transcription O O O activated Pho80 IP4 O X Pho4 O O Pho85 Kinase activity IP receptor blocked O 3 ENDOPLASMIC Pho81 RETICULUM Ca2+ CYTOPLASM NUCLEUS NUCLEUS mRNA export and translation Insulin secretion and AKT Embryonic development Translation termination Effects of IP kinase deficiency O IPMK (IPK2): Multiple defects, death by embryonic day 10 (mice) O O Insulin IPK1: Cillia are shortened and immotile IP6 AKT resistance causing patterning defects (zebrash) O O Multiple defects, death by Ribosome O embryonic day 8.5 (mice) GleI eRF1 Insulin GSK3β Dbp5 ITPK1 (IP56K): Neural tube -

Effect of Enzyme/Substrate Ratio on the Antioxidant Properties of Hydrolysed African Yam Bean
African Journal of Biotechnology Vol. 11(50), pp. 11086-11091, 21 June, 2012 Available online at http://www.academicjournals.org/AJB DOI: 10.5897/AJB11.2271 ISSN 1684–5315 ©2012 Academic Journals Full Length Research Paper Effect of enzyme/substrate ratio on the antioxidant properties of hydrolysed African yam bean Fasasi Olufunmilayo*, Oyebode Esther and Fagbamila Oluwatoyin Department of Food Science and Technology, P. M. B. 704, Federal University of Technology, Akure, Nigeria. Accepted 8 June, 2012 The use of natural antioxidant as compared with synthetic antioxidant in food processing is a growing trend as consumers prefer natural to synthetic antioxidant mainly on emotional ground. This study investigates the antioxidant activity of hydrolysed African yam bean (Sphenostylis sternocarpa) which is regarded as one of the neglected underutilized species (NUS) of crop in Africa and Nigeria especially to improve food security and boost the economic importance of the crop. The antioxidant properties of African yam bean hydrolysates (AYH) produced at different enzyme to substrate (E/S) ratios of 1: 100 and 3: 100 (W/V) using pepsin (pH 2.0, 37°C) were studied. 2, 2-Diphenyl-1-picryl-hydrazyl (DPPH) radical scavenging activity of the hydrolysates was significantly influenced by the E\S ratio as DPPH radical scavenging activity ranged from 56.1 to 75.8% in AYH (1: 100) and 33.3 to 58.8% in AYH (3:100) with 1000 µg/ml having the highest DPPH radical scavenging activity. AYH (1:100) and AYH (3:100) had higher reducing activities of 0.42 and 0.23, respectively at a concentration of 1000 µg/ml. -

Regulation of Energy Substrate Metabolism in Endurance Exercise
International Journal of Environmental Research and Public Health Review Regulation of Energy Substrate Metabolism in Endurance Exercise Abdullah F. Alghannam 1,* , Mazen M. Ghaith 2 and Maha H. Alhussain 3 1 Lifestyle and Health Research Center, Health Sciences Research Center, Princess Nourah bInt. Abdulrahman University, Riyadh 84428, Saudi Arabia 2 Faculty of Applied Medical Sciences, Laboratory Medicine Department, Umm Al-Qura University, Al Abdeyah, Makkah 7607, Saudi Arabia; [email protected] 3 Department of Food Science and Nutrition, College of Food and Agriculture Sciences, King Saud University, Riyadh 11451, Saudi Arabia; [email protected] * Correspondence: [email protected] Abstract: The human body requires energy to function. Adenosine triphosphate (ATP) is the cellular currency for energy-requiring processes including mechanical work (i.e., exercise). ATP used by the cells is ultimately derived from the catabolism of energy substrate molecules—carbohydrates, fat, and protein. In prolonged moderate to high-intensity exercise, there is a delicate interplay between carbohydrate and fat metabolism, and this bioenergetic process is tightly regulated by numerous physiological, nutritional, and environmental factors such as exercise intensity and du- ration, body mass and feeding state. Carbohydrate metabolism is of critical importance during prolonged endurance-type exercise, reflecting the physiological need to regulate glucose homeostasis, assuring optimal glycogen storage, proper muscle fuelling, and delaying the onset of fatigue. Fat metabolism represents a sustainable source of energy to meet energy demands and preserve the ‘limited’ carbohydrate stores. Coordinated neural, hormonal and circulatory events occur during prolonged endurance-type exercise, facilitating the delivery of fatty acids from adipose tissue to the Citation: Alghannam, A.F.; Ghaith, working muscle for oxidation. -

ENZYMES: Catalysis, Kinetics and Mechanisms N
ENZYMES: Catalysis, Kinetics and Mechanisms N. S. Punekar ENZYMES: Catalysis, Kinetics and Mechanisms N. S. Punekar Department of Biosciences & Bioengineering Indian Institute of Technology Bombay Mumbai, Maharashtra, India ISBN 978-981-13-0784-3 ISBN 978-981-13-0785-0 (eBook) https://doi.org/10.1007/978-981-13-0785-0 Library of Congress Control Number: 2018947307 # Springer Nature Singapore Pte Ltd. 2018 This work is subject to copyright. All rights are reserved by the Publisher, whether the whole or part of the material is concerned, specifically the rights of translation, reprinting, reuse of illustrations, recitation, broadcasting, reproduction on microfilms or in any other physical way, and transmission or information storage and retrieval, electronic adaptation, computer software, or by similar or dissimilar methodology now known or hereafter developed. The use of general descriptive names, registered names, trademarks, service marks, etc. in this publication does not imply, even in the absence of a specific statement, that such names are exempt from the relevant protective laws and regulations and therefore free for general use. The publisher, the authors and the editors are safe to assume that the advice and information in this book are believed to be true and accurate at the date of publication. Neither the publisher nor the authors or the editors give a warranty, express or implied, with respect to the material contained herein or for any errors or omissions that may have been made. The publisher remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. Printed on acid-free paper This Springer imprint is published by the registered company Springer Nature Singapore Pte Ltd. -

Phosphoenolpyruvate-Dependent Phosphotransferase System in Lactobacillus Casei BRUCE M
JOURNAL OF BACTERIOLOGY, June 1983, p. 1204-1214 Vol. 154, No. 3 0021-9193/83/061204-11$02.00/0 Copyright C 1983, American Society for Microbiology Regulation and Characterization of the Galactose- Phosphoenolpyruvate-Dependent Phosphotransferase System in Lactobacillus casei BRUCE M. CHASSY* AND JOHN THOMPSON Microbiology Section, Laboratory of Microbiology and Immunology, National Institute of Dental Research, Bethesda, Maryland 20205 Received 8 November 1982/Accepted 5 March 1983 Cells ofLactobacillus casei grown in media containing galactose or a metaboliz- able ,-galactoside (lactose, lactulose, or arabinosyl-P-D-galactoside) were in- duced for a galactose-phosphoenolpyruvate-dependent phosphotransferase sys- tem (gal-PTS). This high-affinity system (Km for galactose, 11 ,uM) was inducible in eight strains examined, which were representative of all five subspecies of L. casei. The gal-PTS was also induced in strains defective in glucose- and lactose- phosphoenolpyruvate-dependent phosphotransferase systems during growth on galactose. Galactose 6-phosphate appeared to be the intracellular inducer of the gal-PTS. The gal-PTS was quite specific for D-galactose, and neither glucose, lactose, nor a variety of structural analogs of galactose caused significant inhibition of phosphotransferase system-mediated galactose transport in intact cells. The phosphoenolpyruvate-dependent phosphorylation of galactose in vitro required specific membrane and cytoplasmic components (including enzyme Illgal), which were induced only by growth of the cells on galactose or ,B- galactosides. Extracts prepared from such cells also contained an ATP-dependent galactokinase which converted galactose to galactose 1-phosphate. Our results demonstrate the separate identities of the gal-PTS and the lactose-phosphoenol- pyruvate-dependent phosphotransferase system in L. -

Determination of Hexokinase and Other Enzymes Which Possibly Phosphorylate Fructose in Neurospora Crassa
Fungal Genetics Reports Volume 8 Article 23 Determination of hexokinase and other enzymes which possibly phosphorylate fructose in Neurospora crassa W. Klingmuller H. G. Truper Follow this and additional works at: https://newprairiepress.org/fgr This work is licensed under a Creative Commons Attribution-Share Alike 4.0 License. Recommended Citation Klingmuller, W., and H.G. Truper (1965) "Determination of hexokinase and other enzymes which possibly phosphorylate fructose in Neurospora crassa," Fungal Genetics Reports: Vol. 8, Article 23. https://doi.org/ 10.4148/1941-4765.2129 This Technical Note is brought to you for free and open access by New Prairie Press. It has been accepted for inclusion in Fungal Genetics Reports by an authorized administrator of New Prairie Press. For more information, please contact [email protected]. Determination of hexokinase and other enzymes which possibly phosphorylate fructose in Neurospora crassa Abstract Determination of hexokinase and other enzymes which possibly phosphorylate fructose in Neurospora crassa This technical note is available in Fungal Genetics Reports: https://newprairiepress.org/fgr/vol8/iss1/23 silky sheen is usually observed when CI suspension of the crystals is agitated; the silky appearance is usually IX)+ present on initial crystallization. It is apparent that an enormous number of variations is possible in carrying out the described procedure. It is therefore of in- terest that each of the twelve systems with which we hove tried this method has allowed crystallization without recourse to changes in pH,temperotwe or other conditions except for the inclusion of a mercaptan where warranted. Ox experience at this time in- cludes dehydrogemses, decarboxylases, transferases and protein hornwnes and involves proteins usucllly sensitive to room tempero- tire, proteins with high and low polysoccharide content and complexes of more than one protein. -

Ceramide Kinase Is Upregulated in Metastatic Breast Cancer Cells and Contributes to Migration and Invasion by Activation of PI 3-Kinase and Akt
International Journal of Molecular Sciences Article Ceramide Kinase Is Upregulated in Metastatic Breast Cancer Cells and Contributes to Migration and Invasion by Activation of PI 3-Kinase and Akt 1,2, 1, 2 2 Stephanie Schwalm y, Martin Erhardt y, Isolde Römer , Josef Pfeilschifter , Uwe Zangemeister-Wittke 1,* and Andrea Huwiler 1,* 1 Institute of Pharmacology, University of Bern, Inselspital, INO-F, CH-3010 Bern, Switzerland; [email protected] (S.S.); [email protected] (M.E.) 2 Institute of General Pharmacology and Toxicology, University Hospital Frankfurt am Main, Goethe-University, Theodor-Stern Kai 7, D-60590 Frankfurt am Main, Germany; [email protected] (I.R.); [email protected] (J.P.) * Correspondence: [email protected] (U.Z.-W.); [email protected] (A.H.); Tel.: +41-31-6323214 (A.H.) These authors contributed equally to this work. y Received: 29 November 2019; Accepted: 12 February 2020; Published: 19 February 2020 Abstract: Ceramide kinase (CerK) is a lipid kinase that converts the proapoptotic ceramide to ceramide 1-phosphate, which has been proposed to have pro-malignant properties and regulate cell responses such as proliferation, migration, and inflammation. We used the parental human breast cancer cell line MDA-MB-231 and two single cell progenies derived from lung and bone metastasis upon injection of the parental cells into immuno-deficient mice. The lung and the bone metastatic cell lines showed a marked upregulation of CerK mRNA and activity when compared to the parental cell line. The metastatic cells also had increased migratory and invasive activity, which was dose-dependently reduced by the selective CerK inhibitor NVP-231. -

Isozymes of Human Phosphofructokinase
Proc. Nat{. Acad. Sci. USA Vol. 77, No. 1, pp. 62-66, January 1980 Biochemistry Isozymes of human phosphofructokinase: Identification and subunit structural characterization of a new system (hemolytic anemia/myopathy/in vitro protein hybridization/column chromatography) SHOBHANA VORA*, CAROL SEAMAN*, SUSAN DURHAM*, AND SERGIO PIOMELLI* Division of Pediatric Hematology, New York University School of Medicine, 550 First Avenue, New York, New York 10016 Communicated by Saul Krugman, July 13, 1979 ABSTRACT The existence of a five-membered isozyme The clinical effects of the enzymatic defect consisted of system for human phosphofructokinase (PFK; ATP:D-fructose- gen- 6-phosphate 1-phosphotransferase, EC 2.7.1.11) has been dem- eralized muscle weakness and compensated hemolysis. The onstrated. These multimolecular forms result from the random differential tissue involvement led to the hypothesis that the polymerization of two distinct subunits, M (muscle type) and erythrocyte isozyme is composed of two types of subunits, one L (liver type), to form all possible tetrameters-i.e., M4, M3L, of which is the sole subunit present in muscle PFK (9, 10). The M2L4, ML3, and L4. Partially purified muscle and liver PFKs proposed structural heterogeneity of erythrocyte PFK protein were hybridized by dissociation at low pH and then recombi- was nation at neutrality. Three hybrid species were generated in supported by immunochemical neutralization experiments addition to the two parental isozymes, to yield an entire five- (11, 12). Karadsheh et al. (13) and Kaur and Layzer (14) have membered set. The various species could be consistently and recently presented data to support the suggested hybrid reproducibly separated from one another by DEAE-Sephadex structure for erythrocyte PFK. -

Spring 2013 Lecture 13-14
CHM333 LECTURE 13 – 14: 2/13 – 15/13 SPRING 2013 Professor Christine Hrycyna INTRODUCTION TO ENZYMES • Enzymes are usually proteins (some RNA) • In general, names end with suffix “ase” • Enzymes are catalysts – increase the rate of a reaction – not consumed by the reaction – act repeatedly to increase the rate of reactions – Enzymes are often very “specific” – promote only 1 particular reaction – Reactants also called “substrates” of enzyme catalyst rate enhancement non-enzymatic (Pd) 102-104 fold enzymatic up to 1020 fold • How much is 1020 fold? catalyst time for reaction yes 1 second no 3 x 1012 years • 3 x 1012 years is 500 times the age of the earth! Carbonic Anhydrase Tissues ! + CO2 +H2O HCO3− +H "Lungs and Kidney 107 rate enhancement Facilitates the transfer of carbon dioxide from tissues to blood and from blood to alveolar air Many enzyme names end in –ase 89 CHM333 LECTURE 13 – 14: 2/13 – 15/13 SPRING 2013 Professor Christine Hrycyna Why Enzymes? • Accelerate and control the rates of vitally important biochemical reactions • Greater reaction specificity • Milder reaction conditions • Capacity for regulation • Enzymes are the agents of metabolic function. • Metabolites have many potential pathways • Enzymes make the desired one most favorable • Enzymes are necessary for life to exist – otherwise reactions would occur too slowly for a metabolizing organis • Enzymes DO NOT change the equilibrium constant of a reaction (accelerates the rates of the forward and reverse reactions equally) • Enzymes DO NOT alter the standard free energy change, (ΔG°) of a reaction 1. ΔG° = amount of energy consumed or liberated in the reaction 2.