Long-Chain 3-Hydroxyacyl-Coa Dehydrogenase Deficiency
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Diapositiva 1
ns 10 A Unmethylated C Methylated ) 8 Normal Normal Tissue Cervix Esophagus 4 cervical esophageal a.u. * *** * *** ( 6 TargetID MAPINFO Position Ca-Ski HeLa SW756 tissue COLO-680N KYSE-180 OACM 5.1C tissue cg04255070 22851591 TSS1500 0.896 0.070 0.082 0.060 0.613 0.622 0.075 0.020 2 (log2) 4 cg01593340 22851587 TSS1500 0.826 0.055 0.051 0.110 0.518 0.518 0.034 0.040 cg16113692 22851502 TSS200 0.964 0.073 0.072 0.030 0.625 0.785 0.018 0.010 2 0 cg26918510 22851422 TSS200 0.985 0.019 0.000 0.000 0.711 0.961 0.010 0.060 expression cg26821579 22851416 TSS200 0.988 0.083 0.058 0.000 0.387 0.955 0.021 0.050 TRMT12 mRNA Expression mRNA TRMT12 0 cg01129966 22851401 TSS200 0.933 0.039 0.077 0.020 0.815 0.963 0.078 0.010 -2 cg25421615 22851383 TSS200 0.958 0.033 0.006 0.000 0.447 0.971 0.037 0.020 mRNA cg17953636 22851321 1stExon 0.816 0.092 0.099 0.080 0.889 0.750 0.136 0.030 -4 Methylated SVIP Unmethylated SVIP relative expression (a.u.) expression relative SVIP D Cervix Esophagus HNC Meth ) HNC Unmeth Cervix Meth 3.0 *** Cervix Unmeth *** Esophagus Meth a.u. ( 2.5 CaCaEsophagus-Ski-Ski Unmeth COLO-680N Haematological Meth TSS B TSS Haematological Unmeth 2.0 -244 bp +104 bp 244 bp +104 bp 1.5 ** * expression -SVIP 1.0 HeLa KYSE-180 HeLa TSS TSS 0.5 -ACTB 244 bp +104 bp - 244 bp +104 bp mRNA 0.0 HeLa SW756 Ca-Ski SW756 SW756 OACM 5.1C TSS KYSE-180 TSS COLO-680NOACM 5.1 C - 244 bp +104 bp 244 bp +104 bp E ) 3.0 * Ca-Ski COLO-680N WT TSS TSS 2.0 a.u. -

Altered Expression and Function of Mitochondrial Я-Oxidation Enzymes
0031-3998/01/5001-0083 PEDIATRIC RESEARCH Vol. 50, No. 1, 2001 Copyright © 2001 International Pediatric Research Foundation, Inc. Printed in U.S.A. Altered Expression and Function of Mitochondrial -Oxidation Enzymes in Juvenile Intrauterine-Growth-Retarded Rat Skeletal Muscle ROBERT H. LANE, DAVID E. KELLEY, VLADIMIR H. RITOV, ANNA E. TSIRKA, AND ELISA M. GRUETZMACHER Department of Pediatrics, UCLA School of Medicine, Mattel Children’s Hospital at UCLA, Los Angeles, California 90095, U.S.A. [R.H.L.]; and Departments of Internal Medicine [D.E.K., V.H.R.] and Pediatrics [R.H.L., A.E.T., E.M.G.], University of Pittsburgh School of Medicine, Magee-Womens Research Institute, Pittsburgh, Pennsylvania 15213, U.S.A. ABSTRACT Uteroplacental insufficiency and subsequent intrauterine creased in IUGR skeletal muscle mitochondria, and isocitrate growth retardation (IUGR) affects postnatal metabolism. In ju- dehydrogenase activity was unchanged. Interestingly, skeletal venile rats, IUGR alters skeletal muscle mitochondrial gene muscle triglycerides were significantly increased in IUGR skel- expression and reduces mitochondrial NADϩ/NADH ratios, both etal muscle. We conclude that uteroplacental insufficiency alters of which affect -oxidation flux. We therefore hypothesized that IUGR skeletal muscle mitochondrial lipid metabolism, and we gene expression and function of mitochondrial -oxidation en- speculate that the changes observed in this study play a role in zymes would be altered in juvenile IUGR skeletal muscle. To test the long-term morbidity associated with IUGR. (Pediatr Res 50: this hypothesis, mRNA levels of five key mitochondrial enzymes 83–90, 2001) (carnitine palmitoyltransferase I, trifunctional protein of -oxi- dation, uncoupling protein-3, isocitrate dehydrogenase, and mi- Abbreviations tochondrial malate dehydrogenase) and intramuscular triglycer- CPTI, carnitine palmitoyltransferase I ides were quantified in 21-d-old (preweaning) IUGR and control IUGR, intrauterine growth retardation rat skeletal muscle. -

Upregulation of Peroxisome Proliferator-Activated Receptor-Α And
Upregulation of peroxisome proliferator-activated receptor-α and the lipid metabolism pathway promotes carcinogenesis of ampullary cancer Chih-Yang Wang, Ying-Jui Chao, Yi-Ling Chen, Tzu-Wen Wang, Nam Nhut Phan, Hui-Ping Hsu, Yan-Shen Shan, Ming-Derg Lai 1 Supplementary Table 1. Demographics and clinical outcomes of five patients with ampullary cancer Time of Tumor Time to Age Differentia survival/ Sex Staging size Morphology Recurrence recurrence Condition (years) tion expired (cm) (months) (months) T2N0, 51 F 211 Polypoid Unknown No -- Survived 193 stage Ib T2N0, 2.41.5 58 F Mixed Good Yes 14 Expired 17 stage Ib 0.6 T3N0, 4.53.5 68 M Polypoid Good No -- Survived 162 stage IIA 1.2 T3N0, 66 M 110.8 Ulcerative Good Yes 64 Expired 227 stage IIA T3N0, 60 M 21.81 Mixed Moderate Yes 5.6 Expired 16.7 stage IIA 2 Supplementary Table 2. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis of an ampullary cancer microarray using the Database for Annotation, Visualization and Integrated Discovery (DAVID). This table contains only pathways with p values that ranged 0.0001~0.05. KEGG Pathway p value Genes Pentose and 1.50E-04 UGT1A6, CRYL1, UGT1A8, AKR1B1, UGT2B11, UGT2A3, glucuronate UGT2B10, UGT2B7, XYLB interconversions Drug metabolism 1.63E-04 CYP3A4, XDH, UGT1A6, CYP3A5, CES2, CYP3A7, UGT1A8, NAT2, UGT2B11, DPYD, UGT2A3, UGT2B10, UGT2B7 Maturity-onset 2.43E-04 HNF1A, HNF4A, SLC2A2, PKLR, NEUROD1, HNF4G, diabetes of the PDX1, NR5A2, NKX2-2 young Starch and sucrose 6.03E-04 GBA3, UGT1A6, G6PC, UGT1A8, ENPP3, MGAM, SI, metabolism -

Role of De Novo Cholesterol Synthesis Enzymes in Cancer Jie Yang1,2, Lihua Wang1,2, Renbing Jia1,2
Journal of Cancer 2020, Vol. 11 1761 Ivyspring International Publisher Journal of Cancer 2020; 11(7): 1761-1767. doi: 10.7150/jca.38598 Review Role of de novo cholesterol synthesis enzymes in cancer Jie Yang1,2, Lihua Wang1,2, Renbing Jia1,2 1. Department of Ophthalmology, Ninth People’s Hospital of Shanghai, Shanghai Jiao Tong University School of Medicine, Shanghai, China. 2. Shanghai Key Laboratory of Orbital Diseases and Ocular Oncology, Shanghai, China. Corresponding authors: Renbing Jia, [email protected] and Lihua Wang, [email protected]. Department of Ophthalmology, Ninth People’s Hospital, Shanghai Jiao Tong University School of Medicine, No. 639 Zhi Zao Ju Road, Shanghai 200011, China © The author(s). This is an open access article distributed under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0/). See http://ivyspring.com/terms for full terms and conditions. Received: 2019.07.21; Accepted: 2019.11.30; Published: 2020.01.17 Abstract Despite extensive research in the cancer field, cancer remains one of the most prevalent diseases. There is an urgent need to identify specific targets that are safe and effective for the treatment of cancer. In recent years, cancer metabolism has come into the spotlight in cancer research. Lipid metabolism, especially cholesterol metabolism, plays a critical role in membrane synthesis as well as lipid signaling in cancer. This review focuses on the contribution of the de novo cholesterol synthesis pathway to tumorigenesis, cancer progression and metastasis. In conclusion, cholesterol metabolism could be an effective target for novel anticancer treatment. Key words: metabolic reprogramming, de novo cholesterol synthesis, cancer progress Introduction Over the past few decades, numerous published that cholesterol plays a critical role in cancer studies have focused on cancer cell metabolism and progression15-19. -

RESEARCH COMMUNICATION HADHA Is a Potential Predictor Of
HADHA is a Potential Predictor of the Response to Platinum-based Chemotherapy RESEARCH COMMUNICATION HADHA is a Potential Predictor of Response to Platinum-based Chemotherapy for Lung Cancer Taihei Kageyama1, Ryo Nagashio1, 2, Shinichiro Ryuge 3, Toshihide Matsumoto1,5, Akira Iyoda4, Yukitoshi Satoh4, Noriyuki Masuda3, Shi-Xu Jiang5, Makoto Saegusa5, Yuichi Sato1, 2* Abstract To identify a cisplatin resistance predictor to reduce or prevent unnecessary side effects, we firstly established four cisplatin-resistant sub-lines and compared their protein profiles with cisplatin-sensitive parent lung cancer cell lines using two-dimensional gel electrophoresis. Between the cisplatin-resistant and -sensitive cells, a total of 359 protein spots were differently expressed (>1.5 fold), and 217 proteins (83.0%) were identified. We focused on a mitochondrial protein, hydroxyl-coenzyme A dehydrogenase/3-ketoacyl-coenzyme A thiolase/enoyl-coenzyme A hydratase alpha subunit (HADHA), which was increased in all cisplatin-resistant cells. Furthermore, pre- treated biopsy specimens taken from patients who showed resistance to platinum-based treatment showed a significantly higher positive rate for HADHA in all cases (p=0.00367), including non-small cell lung carcinomas (p=0.002), small-cell lung carcinomas (p=0.038), and adenocarcinomas (p=0.008). These results suggest that the expression of HADHA may be a useful marker to predict resistance to platinum-based chemotherapy in patients with lung cancer. Keywords: Cisplatin - HADHA - lung cancer - two-dimensional gel electrophoresis Asian Pacific J Cancer Prev, 12, 3457-3463 Introduction cisplatin resistance rose due to a decrease of blood flow in the tumor and increased DNA repair (Stewart, 2007), Lung cancer is the leading cause of cancer-related the mechanisms underlying cisplatin resistance have not death in the world, and the five-year overall survival rate yet been clarified, and an effective cisplatin resistance is still below 16% (Jemal et al., 2009). -

(LCHAD) Deficiency / Mitochondrial Trifunctional Protein (MTF) Deficiency
Long chain acyl-CoA dehydrogenase (LCHAD) deficiency / Mitochondrial trifunctional protein (MTF) deficiency Contact details Introduction Regional Genetics Service Long chain acyl-CoA dehydrogenase (LCHAD) deficiency / mitochondrial trifunctional Levels 4-6, Barclay House protein (MTF) deficiency is an autosomal recessive disorder of mitochondrial beta- 37 Queen Square oxidation of fatty acids. The mitochondrial trifunctional protein is composed of 4 alpha London, WC1N 3BH and 4 beta subunits, which are encoded by the HADHA and HADHB genes, respectively. It is characterized by early-onset cardiomyopathy, hypoglycemia, T +44 (0) 20 7762 6888 neuropathy, and pigmentary retinopathy, and sudden death. There is also an infantile F +44 (0) 20 7813 8578 onset form with a hepatic Reye-like syndrome, and a late-adolescent onset form with primarily a skeletal myopathy. Tandem mass spectrometry of organic acids in urine, Samples required and carnitines in blood spots, allows the diagnosis to be unequivocally determined. An 5ml venous blood in plastic EDTA additional clinical complication can occur in the pregnant mothers of affected fetuses; bottles (>1ml from neonates) they may experience maternal acute fatty liver of pregnancy (AFLP) syndrome or Prenatal testing must be arranged hypertension/haemolysis, elevated liver enzymes and low platelets (HELLP) in advance, through a Clinical syndrome. Genetics department if possible. The genes encoding the HADHA and HADHB subunits are located on chromosome Amniotic fluid or CV samples 2p23.3. The pathogenic -

Lipid Metabolic Reprogramming: Role in Melanoma Progression and Therapeutic Perspectives
cancers Review Lipid metabolic Reprogramming: Role in Melanoma Progression and Therapeutic Perspectives 1, 1, 1 2 1 Laurence Pellerin y, Lorry Carrié y , Carine Dufau , Laurence Nieto , Bruno Ségui , 1,3 1, , 1, , Thierry Levade , Joëlle Riond * z and Nathalie Andrieu-Abadie * z 1 Centre de Recherches en Cancérologie de Toulouse, Equipe Labellisée Fondation ARC, Université Fédérale de Toulouse Midi-Pyrénées, Université Toulouse III Paul-Sabatier, Inserm 1037, 2 avenue Hubert Curien, tgrCS 53717, 31037 Toulouse CEDEX 1, France; [email protected] (L.P.); [email protected] (L.C.); [email protected] (C.D.); [email protected] (B.S.); [email protected] (T.L.) 2 Institut de Pharmacologie et de Biologie Structurale, CNRS, Université Toulouse III Paul-Sabatier, UMR 5089, 205 Route de Narbonne, 31400 Toulouse, France; [email protected] 3 Laboratoire de Biochimie Métabolique, CHU Toulouse, 31059 Toulouse, France * Correspondence: [email protected] (J.R.); [email protected] (N.A.-A.); Tel.: +33-582-7416-20 (J.R.) These authors contributed equally to this work. y These authors jointly supervised this work. z Received: 15 September 2020; Accepted: 23 October 2020; Published: 27 October 2020 Simple Summary: Melanoma is a devastating skin cancer characterized by an impressive metabolic plasticity. Melanoma cells are able to adapt to the tumor microenvironment by using a variety of fuels that contribute to tumor growth and progression. In this review, the authors summarize the contribution of the lipid metabolic network in melanoma plasticity and aggressiveness, with a particular attention to specific lipid classes such as glycerophospholipids, sphingolipids, sterols and eicosanoids. -

Supplementary Materials
Supplementary Materials COMPARATIVE ANALYSIS OF THE TRANSCRIPTOME, PROTEOME AND miRNA PROFILE OF KUPFFER CELLS AND MONOCYTES Andrey Elchaninov1,3*, Anastasiya Lokhonina1,3, Maria Nikitina2, Polina Vishnyakova1,3, Andrey Makarov1, Irina Arutyunyan1, Anastasiya Poltavets1, Evgeniya Kananykhina2, Sergey Kovalchuk4, Evgeny Karpulevich5,6, Galina Bolshakova2, Gennady Sukhikh1, Timur Fatkhudinov2,3 1 Laboratory of Regenerative Medicine, National Medical Research Center for Obstetrics, Gynecology and Perinatology Named after Academician V.I. Kulakov of Ministry of Healthcare of Russian Federation, Moscow, Russia 2 Laboratory of Growth and Development, Scientific Research Institute of Human Morphology, Moscow, Russia 3 Histology Department, Medical Institute, Peoples' Friendship University of Russia, Moscow, Russia 4 Laboratory of Bioinformatic methods for Combinatorial Chemistry and Biology, Shemyakin-Ovchinnikov Institute of Bioorganic Chemistry of the Russian Academy of Sciences, Moscow, Russia 5 Information Systems Department, Ivannikov Institute for System Programming of the Russian Academy of Sciences, Moscow, Russia 6 Genome Engineering Laboratory, Moscow Institute of Physics and Technology, Dolgoprudny, Moscow Region, Russia Figure S1. Flow cytometry analysis of unsorted blood sample. Representative forward, side scattering and histogram are shown. The proportions of negative cells were determined in relation to the isotype controls. The percentages of positive cells are indicated. The blue curve corresponds to the isotype control. Figure S2. Flow cytometry analysis of unsorted liver stromal cells. Representative forward, side scattering and histogram are shown. The proportions of negative cells were determined in relation to the isotype controls. The percentages of positive cells are indicated. The blue curve corresponds to the isotype control. Figure S3. MiRNAs expression analysis in monocytes and Kupffer cells. Full-length of heatmaps are presented. -
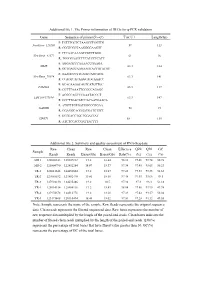
Additional File 1
Additional file 1. The Primer information of DEGs for q-PCR validation Gene Sequence of primer(5'→3') Tm(℃) Length(bp) F: TGTTTGCTCTAAGCCTGGTTG NewGene_126260 59 113 R: CGGTCGCTAAGGGGAAGTT F: CTCAACAAAGCCGTCTGGG NewGene_41572 61 96 R: TGGGGAATCTTCATCCTCATT F: AGGAGCCCAAAACCGAAGA MME 63.3 184 R: GCTGACCAAGAAGTACCGTATGT F: AAAGCCCTTCAGTCAGCACG NewGene_70974 63.3 141 R: CCAGTCACAAGCAGCAAACC F: GCACAAGGCAGTCATGTTGC FAM43A 63.3 117 R: CGTTTAAATTCCGCCAGAGC F: ACGGCAGCCCAAATACCCT LOC108177184 63.3 147 R: GCCTTGACATCCACAATGAACA F: ATGTTTGTGATGGGCGTGAA GAPDH 58 94 R: GGAGGCAGGGATGATGTTCT F: GCTGACCTGCTGGATTAT HPRT1 58 135 R: ATCTCCACCGATTACTTT Additional file 2. Summary and quality assessment of RNA-Seq data Raw Clean Raw Clean Effective Q20 Q30 GC Sample Reads Reads Bases(Gb) Bases(Gb) Rate(%) (%) (%) (%) MR-1 128008342 125607312 19.2 18.84 98.12 97.46 93.74 50.78 MR-2 125804790 122452284 18.87 18.37 97.34 97.45 93.65 50.23 TR-1 128011648 124452604 19.2 18.67 97.22 97.33 93.35 51.65 TR-2 127085532 123882470 19.06 18.58 97.48 97.45 93.66 49.5 TR-3 127994130 124635486 19.2 18.7 97.38 97.3 93.3 51.18 YR-1 128014314 125504116 19.2 18.83 98.04 97.56 93.95 49.98 YR-2 127974678 124512778 19.2 18.68 97.29 97.42 93.57 50.04 YR-3 123173408 120110494 18.48 18.02 97.51 97.28 93.32 49.55 Note: Sample represents the name of the sample. Raw Reads represents the original sequence data. Clean reads represents the filtered sequenced data. Raw bases represents the number of raw sequence data multiplied by the length of the paired-end reads. -

And NADPH-Driven Reactions of Mitochondrial Isocitrate
www.nature.com/scientificreports OPEN SIRT3 and GCN5L regulation of NADP+- and NADPH-driven reactions of mitochondrial isocitrate dehydrogenase IDH2 Katarína Smolková 1 ✉ , Jitka Špačková1, Klára Gotvaldová1, Aleš Dvořák1,4, Alena Křenková2, Martin Hubálek2, Blanka Holendová 1, Libor Vítek3 & Petr Ježek1 Wild type mitochondrial isocitrate dehydrogenase (IDH2) was previously reported to produce oncometabolite 2-hydroxyglutarate (2HG). Besides, mitochondrial deacetylase SIRT3 has been shown to regulate the oxidative function of IDH2. However, regulation of 2HG formation by SIRT3- mediated deacetylation was not investigated yet. We aimed to study mitochondrial IDH2 function in response to acetylation and deacetylation, and focus specifcally on 2HG production by IDH2. We used acetylation surrogate mutant of IDH2 K413Q and assayed enzyme kinetics of oxidative decarboxylation of isocitrate, 2HG production by the enzyme, and 2HG production in cells. The purifed IDH2 K413Q exhibited lower oxidative reaction rates than IDH2 WT. 2HG production by IDH2 K413Q was largely diminished at the enzymatic and cellular level, and knockdown of SIRT3 also inhibited 2HG production by IDH2. Contrary, the expression of putative mitochondrial acetylase GCN5L likely does not target IDH2. Using mass spectroscopy, we further identifed lysine residues within IDH2, which are the substrates of SIRT3. In summary, we demonstrate that 2HG levels arise from non-mutant IDH2 reductive function and decrease with increasing acetylation level. The newly identifed lysine residues might apply in regulation of IDH2 function in response to metabolic perturbations occurring in cancer cells, such as glucose-free conditions. Lysine acylation is a reversible post-translational modification representing one of the primary regulatory mechanisms in mitochondria, typically inhibition of protein function, and includes acetylation, malonylation, succinylation, glutarylation, etc.1. -

Metabolomics-Assisted Proteomics Identifies Succinylation and SIRT5 As Important Regulators of Cardiac Function
Metabolomics-assisted proteomics identifies succinylation and SIRT5 as important regulators of cardiac function Sushabhan Sadhukhana, Xiaojing Liub,c,d, Dongryeol Ryue, Ornella D. Nelsona, John A. Stupinskif, Zhi Lia, Wei Cheng, Sheng Zhangg, Robert S. Weissf, Jason W. Locasaleb,c,d, Johan Auwerxe,1, and Hening Lina,h,1 aDepartment of Chemistry and Chemical Biology, Cornell University, Ithaca, NY 14853; bDuke Cancer Institute, Duke University School of Medicine, Durham, NC 27710; cDuke Molecular Physiology Institute, Duke University School of Medicine, Durham, NC 27710; dDepartment of Pharmacology and Cancer Biology, Duke University School of Medicine, Durham, NC 27710; eLaboratory of Integrative and Systems Physiology, School of Life Sciences, École Polytechnique Fédérale de Lausanne, 1015 Lausanne, Switzerland; fDepartment of Biomedical Sciences, Cornell University, Ithaca, NY 14853; gProteomics & Mass Spectrometry Facility, Institute of Biotechnology, Cornell University, Ithaca, NY 14853; and hHoward Hughes Medical Institute, Cornell University, Ithaca, NY 14853 Edited by Kevan M. Shokat, University of California, San Francisco, CA, and approved March 9, 2016 (received for review October 7, 2015) Cellular metabolites, such as acyl-CoA, can modify proteins, leading SIRT4 and SIRT5 have very weak deacetylase activities (14). SIRT5 to protein posttranslational modifications (PTMs). One such PTM is possesses unique enzymatic activity on hydrolyzing negatively lysine succinylation, which is regulated by sirtuin 5 (SIRT5). Although charged -

Epigenetic Mechanisms Are Involved in the Oncogenic Properties of ZNF518B in Colorectal Cancer
Epigenetic mechanisms are involved in the oncogenic properties of ZNF518B in colorectal cancer Francisco Gimeno-Valiente, Ángela L. Riffo-Campos, Luis Torres, Noelia Tarazona, Valentina Gambardella, Andrés Cervantes, Gerardo López-Rodas, Luis Franco and Josefa Castillo SUPPLEMENTARY METHODS 1. Selection of genomic sequences for ChIP analysis To select the sequences for ChIP analysis in the five putative target genes, namely, PADI3, ZDHHC2, RGS4, EFNA5 and KAT2B, the genomic region corresponding to the gene was downloaded from Ensembl. Then, zoom was applied to see in detail the promoter, enhancers and regulatory sequences. The details for HCT116 cells were then recovered and the target sequences for factor binding examined. Obviously, there are not data for ZNF518B, but special attention was paid to the target sequences of other zinc-finger containing factors. Finally, the regions that may putatively bind ZNF518B were selected and primers defining amplicons spanning such sequences were searched out. Supplementary Figure S3 gives the location of the amplicons used in each gene. 2. Obtaining the raw data and generating the BAM files for in silico analysis of the effects of EHMT2 and EZH2 silencing The data of siEZH2 (SRR6384524), siG9a (SRR6384526) and siNon-target (SRR6384521) in HCT116 cell line, were downloaded from SRA (Bioproject PRJNA422822, https://www.ncbi. nlm.nih.gov/bioproject/), using SRA-tolkit (https://ncbi.github.io/sra-tools/). All data correspond to RNAseq single end. doBasics = TRUE doAll = FALSE $ fastq-dump -I --split-files SRR6384524 Data quality was checked using the software fastqc (https://www.bioinformatics.babraham. ac.uk /projects/fastqc/). The first low quality removing nucleotides were removed using FASTX- Toolkit (http://hannonlab.cshl.edu/fastxtoolkit/).