Development of an Effective LC-MS/MS Cleaning Validation Method for Synthetic Peptide Drug Substances
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

VITAL Jan 2019 Follow-Up
VITAL 6308 Request Use ball-point pen to complete the form. OBS 1 DATE OF BIRTH: / / We use DATE OF BIRTH (DOB) to verify the identity of the person providing information. Is the DOB above correct? Yes No IF NO, what is your correct date of birth? / / s. Peripheral artery surgery / 1. IN THE PAST YEAR, have you been NEWLY DIAGNOSED stenting (procedure to No Yes / with any of the following? IF YES, please provide the unblock arteries in legs) month/year of the NEW diagnosis or procedure. t. Carotid stenosis (blocked No Yes Answer NO/YES on each line. arteries in neck) / Diagnosis (Please complete either N/Y for each item) MO/YR u. Carotid artery surgery / stenting (procedure to No Yes / a. Hypertension (high blood pressure) No Yes / unblock arteries in neck) v. Deep vein thrombosis No Yes (blood clot in legs) / b. Diabetes No Yes / w. Pulmonary embolism No Yes (blood clot in lungs) / c. Cancer (NOT including skin cancer) No Yes / IF YES, specify type: _______________________ x. Parkinson's disease No Yes / d. Skin cancer No Yes / y. Multiple sclerosis No Yes IF YES, specify type: / e. melanoma squamous or basal cell not sure z. Cataract surgery (extraction) No Yes / f. Heart attack or myocardial infarction No Yes / aa. Macular degeneration No Yes / g. Coronary bypass surgery No Yes / bb. Dry eye syndrome No Yes or dry eye disease / h. Coronary angioplasty or stent No Yes (balloon used to unblock an artery) / cc. Periodontal disease No Yes (gum disease) / i. Chest pain (angina) No Yes / IF YES, were you hospitalized? No Yes dd. -

MSM Chapter 1200 3/1/21
MEDICAID SERVICES MANUAL TRANSMITTAL LETTER February 23, 2021 TO: CUSTODIANS OF MEDICAID SERVICES MANUAL FROM: JESSICA KEMMERER, HIPAA PRIVACY AND CIVIL RIGHTS OFFICER /Jessica Kemmerer/ BACKGROUND AND EXPLANATION The DHCFP is proposing revisions to Medicaid Services Manual (MSM), Chapter 1200 – Prescribed Drugs, Appendix A, to reflect recommendations approved on October 22, 2020, by the Drug Use Review (DUR) Board. The proposed changes include the addition of new prior authorization criteria for Doxepine Topical, the addition of new prior authorization criteria for Zeposia® (ozanimod), addition of new prior authorization for Evenity® (romosozumab-aqqg), Prolia® (denosumab), Forteo® (teriparatide) and Tymlos® (abaloparatide) within a new combined osteoporosis agents section, and addition of new prior authorization criteria for Orilissa® (elagolix) and Oriahnn® (elagolix, estradiol, and norethindrone) within a new Gonadorpin Hormone Receptor (GnRH) Antagonist and Combinations section. Additionally, the DHCFP is proposing revisions to the existing prior authorization criteria for psychotropic medications for children and adolescents, and revision to the existing clinical criteria for Epidiolex® (cannabidiol). Throughout the chapter, grammar, punctuation and capitalization changes were made, duplications removed, acronyms used and standardized, and language reworded for clarity. Renumbering and re- arranging of sections was necessary. These changes are effective March 1, 2021. MATERIAL TRANSMITTED MATERIAL SUPERSEDED MTL N/A MTL N/A MSM Ch 1200 – Prescribed Drugs MSM Ch 1200 – Prescribed Drugs Background and Explanation of Policy Changes, Manual Section Section Title Clarifications and Updates Appendix A Psychotropic Added new policy language criteria on which specific Section N Medications for drug classes may bypass polypharmacy clinical criteria. Children and Adolescents Appendix A Reserved for Future Created a new section titled “Doxepin Topical.” Added Section W Use new prior authorization criteria for doxepin topical. -

2017 Fda Peptide Harvest
Preprints (www.preprints.org) | NOT PEER-REVIEWED | Posted: 10 April 2018 doi:10.20944/preprints201804.0126.v1 Peer-reviewed version available at Pharmaceuticals 2018, 11, 42; doi:10.3390/ph11020042 1 Review 2 2017 FDA PEPTIDE HARVEST 3 Othman Al Musaimi,1,2,# Danah Alshaer, 1,2,# Beatriz G. de la Torre,3,* Fernando Albericio,2,4,5.* 4 1 College of Health Sciences, University of KwaZulu-Natal, Durban 4000, South Africa 5 2 School of Chemistry, University of KwaZulu-Natal, Durban 4001, South Africa 6 3 KRISP, College of Health Sciences, University of KwaZulu-Natal, Durban 4001, South Africa 7 4 CIBER-BBN, Networking Centre on Bioengineering, Biomaterials and Nanomedicine, University of 8 Barcelona, 08028 Barcelona, Spain 9 5 Department of Organic Chemistry, University of Barcelona, 08028 Barcelona, Spain 10 * Correspondence: [email protected]; [email protected]; Tel.: +27-614009144 11 12 13 Abstract: 2017 was an excellent year in terms of new drugs (chemical entities and biologics) 14 approved by the FDA, with a total of forty-six. In turn, one of the highlights was the number of 15 peptides (six) included in this list. Here, the six peptides are analysed in terms of chemical structure, 16 synthetic strategy used for their production, source, biological target, and mode of action. 17 Keywords: pharmaceutical market; drugs; drug discovery; solid-phase peptide synthesis 18 Introduction 19 The financial investment associated with the pharmaceutical industry is one of the largest in the 20 industrial sector—surpassed only by the telecommunications sector. However, the number of new 21 products (drugs) entering the market each year is relatively low. -

Medicaid / CHP+ Prior Authorization Criteria
Prior Authorization Approval Criteria Effective Date: 07/01/2021 Prior authorization criteria is developed following evidence-based criteria including: i. Safety, including concurrent drug utilization review (cDUR) when applicable ii. Efficacy: the potential outcome of treatment under optimal circumstances iii. Strength of scientific evidence and standards of practice through review of relevant information from the peer-reviewed medical literature, accepted national treatment guidelines, and expert opinion where necessary iv. Cost-Effectiveness: the actual outcome of treatment under real life conditions including consideration of total health care costs, not just drug costs, through utilization of pharmacoeconomic principles and/or published pharmacoeconomic or outcomes research evaluations where available v. Relevant benefits of current formulary agents of similar use vi. Any restrictions that should be delineated to assure safe, effective, or proper use of the drug. Page 1 of 3 This document contains Prior Authorization Approval Criteria for the following medications: 1. Abilify Maintena (aripiprazole long-acting injectable) 2. Aimovig (erenumab) 3. Ajovy (fremanezumab) 4. Ampyra (dalfampridine) 5. Aubagio (teriflunomide) 6. Briviact (brivaracetam) 7. Cimzia (certolizumab) 8. Cosentyx (secukinumab) 9. Cuvposa (glycopyrrolate oral solution) 10. Daytrana (methylphenidate extended release transdermal system) 11. Dupixent (dupilumab) 12. Emgality (galcanezumab) 13. Epidiolex (cannibidiol) 14. Fanapt (iloperidone) 15. Fetzima (levomilnacipran) 16. Forteo (teriparatide) 17. Gilenya (fingolimod) 18. Gleevec (imatinib) 19. Hepatitis C Virus (HCV) Non-Preferred Medications (Mavyret, Zepatier) 20. Hepatitis C Virus (HCV) Preferred Medications (Epclusa, Harvoni) 21. Horizant (gabapentin enacarbil) 22. Invega (paliperidone) 23. Invega Sustenna (paliperidone palmitate) 24. Jakafi (ruxolitinib) 25. Kalydeco (ivacaftor) 26. Kapvay (clonidine extended release) 27. Latuda (lurasidone) 28. Lupron, Lupron Depot, Lupron Depot-Ped (leuprolide) 29. -

Abaloparatide
ABALOPARATIDE Products Affected • TYMLOS PA Criteria Criteria Details Exclusion Criteria Required Medical ONE OF THE FOLLOWING: (1) HIGH RISK FOR FRACTURES Information DEFINED AS ONE OF THE FOLLOWING: HISTORY OF OSTEOPOROTIC (I.E., FRAGILITY, LOW TRAUMA) FRACTURE(S). 2 OR MORE RISK FACTORS FOR FRACTURE (E.G., HISTORY OF MULTIPLE RECENT LOW TRAUMA FRACTURES, BMD T-SCORE LESS THAN OR EQUAL TO -2.5, CORTICOSTEROID USE, OR USE OF GNRH ANALOGS SUCH AS NAFARELIN, ETC.). NO PRIOR TREATMENT FOR OSTEOPOROSIS AND FRAX SCORE OF AT LEAST 20% FOR ANY MAJOR FRACTURE OR OF AT LEAST 3% FOR HIP FRACTURE. (2) UNABLE TO USE ORAL THERAPY (I.E., UPPER GASTROINTESTINAL PROBLEMS UNABLE TO TOLERATE ORAL MEDICATION, LOWER GASTROINTESTINAL PROBLEMS UNABLE TO ABSORB ORAL MEDICATIONS, TROUBLE REMEMBERING TO TAKE ORAL MEDICATIONS OR COORDINATING AN ORAL BISPHOSPHONATE WITH OTHER ORAL MEDICATIONS OR THEIR DAILY ROUTINE). (3) ADEQUATE TRIAL OF, INTOLERANCE TO, OR A CONTRAINDICATION TO BISPHOSPHONATES (E.G., FOSAMAX, ACTONEL, BONIVA). Age Restrictions Prescriber Restrictions Coverage 12 MONTHS Duration Other Criteria 1 PA Criteria Criteria Details Indications All FDA-approved Indications. Off Label Uses 2 ABEMACICLIB Products Affected • VERZENIO PA Criteria Criteria Details Exclusion Criteria Required Medical Information Age Restrictions Prescriber Restrictions Coverage 12 MONTHS Duration Other Criteria Indications All FDA-approved Indications. Off Label Uses 3 ACALABRUTINIB Products Affected • CALQUENCE PA Criteria Criteria Details Exclusion Criteria Required Medical Information Age Restrictions Prescriber Restrictions Coverage 12 MONTHS Duration Other Criteria Indications All FDA-approved Indications. Off Label Uses 4 ACTEMRA IV (S) Products Affected • ACTEMRA PA Criteria Criteria Details Exclusion ACTIVE SERIOUS INFECTION (INCLUDING Criteria TUBERCULOSIS). -

Talking with Nobel Laureate Robert J. Lefkowitz, MD
MEET THE 2021 ENDOCRINE SOCIETY LAUREATE AWARD WINNERS APRIL 2021 THE LEADING MAGAZINE FOR ENDOCRINOLOGISTS INTERNATIONAL The Accidental SCIENTIST Talking with Nobel Laureate Robert J. Lefkowitz, MD When Robert J. Lefkowitz, MD, received the Nobel Prize for Chemistry in 2012, that was only part of his storied career as a physician scientist. A self-proclaimed “accidental scientist,” Lefkowitz talks to Endocrine News about his recently published autobiography, misconceptions about scientists, how it felt to win the Nobel, and much more! ON THE MOVE: Joy Wu, MD, PhD, relocates her lab during a pandemic. STAR POWER: Rounding up the Rising Stars Power Talks winners CLINICAL PRACTICE GUIDELINES FROM THE ENDOCRINE SOCIETY THE LEADING MAGAZINE FOR ENDOCRINOLOGISTS 2020 – 2022 EDITORIAL ADVISORY BOARD Henry Anhalt, DO Bergen County Pediatric Endocrinology Sally Camper, PhD Department of Human Genetics University of Michigan Medical School Rodolfo J. Galindo, MD Assistant Professor of Medicine Mount Sinai School of Medicine Christian M. Girgis, MBBS, PhD, FRACP Royal North Shore and Westmead Hospitals University of Sydney, Australia Lipid Management Andrea Gore, PhD Division of Pharmacology and Toxicology University of Texas in Patients with Daniel A. Gorelick, PhD Endocrine Disorders Baylor University, Houston, Texas M. Carol Greenlee, MD, FACP A NEW STANDARD FOR CARE Western Slope Endocrinology Grand Junction, Colo. (Faculty for Transforming Clinical Practice initiative [TCPi]) Learn the latest best practices Gary D. Hammer, MD, PhD Millie Schembechler Professor of Adrenal Cancer, for assessing and treating high Endocrine Oncology Program cholesterol in patients with endocrine University of Michigan diseases like hypothyroidism, Robert W. Lash, MD Chief Professional & Clinical Officer, Endocrine Society menopause, and Cushing Syndrome. -

Strides/Stelis Investor Day 2019
Advancing Affordable Biopharma Stelis Leadership Team today Milan Doshi Dr. Roger Lias SVP & Global Biosimilars CEO Business Development Joe Thomas Biju Mathew Outgoing CEO SVP & Head of Quality Minh Tran Molly McGlaughlin SVP & Head of Manufacturing SVP & Global Lead CDMO Anand Khedkar Sachin Jaiswal SVP & Head of R&D Finance Head page Strictly Private and Confidential 02 EVOLUTION. Compelling opportunity Differentiated Model $160m+ investments Strong progress What started as a sub-set of our speciality business has now been built into a fully integrated biopharmaceutical business with world-class capabilities. Biologics is a long gestation business and Stelis is at the end of the investment phase with products de- risked & close to filing and commercial-scale GMP facilities coming on stream to deliver revenues Strictly Private and Confidential Invested over $160m to position Stelis as a differentiated business having diverse income streams UNIQUE SCIENTIFIC DOMAIN NICHE DIVERSIFIED PLATFORM EXPERIENCE EXPERTISE PORTFOLIO INCOME STREAMS 01 02 03 04 05 State-of-the art R&D and small Talented scientific & Clone development Focusing on Bone health Near term revenues from scale manufacturing facility technical team with Cell culture and and Diabetes CDMO/CMO income to service focusing on biosimilars, bio- experience in high-end fermentation operating costs at Stelis betters and novel biologic biopharma development Process engineering and 2 assets ready to enter applications & manufacturing to analytics phase-3 clinical study Phased -

Abaloparatide
ABALOPARATIDE Products Affected • TYMLOS PA Criteria Criteria Details Covered Uses ALL FDA APPROVED INDICATIONS NOT OTHERWISE EXCLUDED FROM PART D. Exclusion Criteria Required Medical ONE OF THE FOLLOWING: (1) HIGH RISK FOR FRACTURES Information DEFINED AS ONE OF THE FOLLOWING: HISTORY OF OSTEOPOROTIC (I.E., FRAGILITY, LOW TRAUMA) FRACTURE(S). 2 OR MORE RISK FACTORS FOR FRACTURE (E.G., HISTORY OF MULTIPLE RECENT LOW TRAUMA FRACTURES, BMD T-SCORE LESS THAN OR EQUAL TO -2.5, CORTICOSTEROID USE, OR USE OF GNRH ANALOGS SUCH AS NAFARELIN, ETC.). NO PRIOR TREATMENT FOR OSTEOPOROSIS AND FRAX SCORE OF AT LEAST 20% FOR ANY MAJOR FRACTURE OR OF AT LEAST 3% FOR HIP FRACTURE. (2) UNABLE TO USE ORAL THERAPY (I.E., UPPER GASTROINTESTINAL PROBLEMS UNABLE TO TOLERATE ORAL MEDICATION, LOWER GASTROINTESTINAL PROBLEMS UNABLE TO ABSORB ORAL MEDICATIONS, TROUBLE REMEMBERING TO TAKE ORAL MEDICATIONS OR COORDINATING AN ORAL BISPHOSPHONATE WITH OTHER ORAL MEDICATIONS OR THEIR DAILY ROUTINE). (3) ADEQUATE TRIAL OF, INTOLERANCE TO, OR A CONTRAINDICATION TO BISPHOSPHONATES (E.G., ALENDRONATE, RISEDRONATE, IBANDRONATE). Age Restrictions Prescriber Restrictions 1 PA Criteria Criteria Details Coverage 12 MONTHS Duration Other Criteria 2 ABATACEPT IV Products Affected • ORENCIA (WITH MALTOSE) PA Criteria Criteria Details Covered Uses ALL FDA APPROVED INDICATIONS NOT OTHERWISE EXCLUDED FROM PART D. Exclusion Criteria Required Medical RENEWAL: PHYSICIAN ATTESTATION OF IMPROVEMENT. Information Age Restrictions Prescriber RHEUMATOID ARTHRITIS, JUVENILE IDIOPATHIC Restrictions ARTHRITIS: PRESCRIBED BY OR GIVEN IN CONSULTATION WITH A RHEUMATOLOGIST. PSORIATIC ARTHRITIS: PRESCRIBED BY OR GIVEN IN CONSULTATION WITH A DERMATOLOGIST OR RHEUMATOLOGIST. Coverage INITIAL: RA: 6 MOS. JIA: 4 MOS. PSA: 12 MOS. -

1 Trends in Peptide Therapeutics
3 1 Trends in Peptide Therapeutics Florence M. Brunel1, Fa Liu2, and John P. Mayer3 1 Novo‐Nordisk Research Center, 5225 Exploration Dr., Indianapolis, IN, 46241, USA 2 Novo‐Nordisk Research Center, 530 Fairview Avenue North, Seattle, WA, 98109, USA 3 University of Colorado, MCD Biology, 1945 Colorado Avenue, Boulder, CO, 80309, USA 1.1 Introduction The growing importance of peptide drugs within the pharmacopoeia has become evident over the past several decades. Among the factors that have contributed to this trend is the recognition that peptide ligands regulate a multitude of physi- ological pathways and are often suitable for therapeutic applications, in either their native or modified form. In addition, certain attributes that are unique to peptides, such as their high selectivity, potency, and lack of toxicity, have ulti- mately become appreciated. The alternative means of drugging peptide recep- tors through target‐directed screening or rational design of orally available small molecules have, with few exceptions, proved unproductive. Mimicking the activ- ity of a peptide agonist is highly challenging, particularly in the case of Class II G‐protein‐coupled receptor (GPCR) targets. Successful examples have typically involved receptor antagonists such as neurokinin, angiotensin, endothelin, and orexin. These lessons have increasingly led drug discovery scientists to consider peptides as legitimate drug candidates, rather than leads or proof‐of‐concept models for small‐molecule programs. Peptide medicinal chemists have also had to confront and overcome shortcomings such as rapid metabolism, clearance, production costs, and limited alternative delivery options. In the present chapter, we highlight the role of peptides in therapeutic areas such as metabolic disease, where peptides have been well established, as well as in areas where their impact has been minor, but now rapidly expanding. -

The Impact of Antiosteoporotic Drugs on Glucose Metabolism and Fracture Risk in Diabetes: Good Or Bad News?
Journal of Clinical Medicine Review The Impact of Antiosteoporotic Drugs on Glucose Metabolism and Fracture Risk in Diabetes: Good or Bad News? Athanasios D. Anastasilakis 1,† , Elena Tsourdi 2,*,†, Gaia Tabacco 3 , Anda Mihaela Naciu 3 , Nicola Napoli 3, Fabio Vescini 4,‡ and Andrea Palermo 3,‡ 1 Department of Endocrinology, 424 General Military Hospital, 56429 Thessaloniki, Greece; [email protected] 2 Department of Medicine (III) &Center for Healthy Aging, Technische Universität Dresden Medical Center, 01307 Dresden, Germany 3 Unit of Endocrinology and Diabetes, Campus Bio-Medico University, 00128 Rome, Italy; [email protected] (G.T.); [email protected] (A.M.N.); [email protected] (N.N.); [email protected] (A.P.) 4 Department of Endocrinology and Diabetes, Santa Maria della Misericordia Hospital, 33100 Udine, Italy; [email protected] * Correspondence: [email protected]; Tel.: +49-351-458-12933; Fax: +49-351-458-5801 † These authors contributed equally to this manuscript. ‡ These authors contributed equally to this manuscript. Abstract: Osteoporosis and diabetes mellitus represent global health problems due to their high, and increasing with aging, prevalence in the general population. Osteoporosis can be successfully treated with both antiresorptive and anabolic drugs. While these drugs are clearly effective in reducing the risk of fracture in patients with postmenopausal and male osteoporosis, it is still unclear whether they may have the same efficacy in patients with diabetic osteopathy. Furthermore, as bone-derived Citation: Anastasilakis, A.D.; cytokines (osteokines) are able to influence glucose metabolism, it is conceivable that antiosteoporotic Tsourdi, E.; Tabacco, G.; Naciu, A.M.; drugs may have an effect on glycemic control through their modulation of bone turnover that Napoli, N.; Vescini, F.; Palermo, A. -
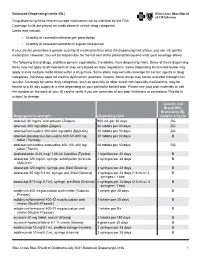
Balanced Drug List Dispensing Limits
Balanced Dispensing Limits (DL) Drug dispensing limits help encourage medication use as intended by the FDA. Coverage limits are placed on medications in certain drug categories. Limits may include: • Quantity of covered medication per prescription • Quantity of covered medication in a given time period If your doctor prescribes a greater quantity of medication than what the dispensing limit allows, you can still get the medication. However, you will be responsible for the full cost of the prescription beyond what your coverage allows. The following brand drugs, and their generic equivalents, if available, have dispensing limits. Some of these dispensing limits may not apply to all members or may vary based on state regulations. Some dispensing limits listed below may apply across multiple medications within a drug class. Some plans may exclude coverage for certain agents or drug categories, like those used for erectile dysfunction (example: Viagra). Some drugs may not be available through mail service. Coverage for some drug categories, such as specialty or other select non‑specialty medications, may be limited to a 30‑day supply at a time depending on your particular benefit plan. Please see your plan materials or call the number on the back of your ID card to verify if you are uncertain of any plan limitations or exclusions. This list is subject to change. Generic and Brand (BG), Brand Only (B), Drug (generic) strength Dispensing Limit Generic only (G) abacavir 20 mg/mL oral solution (Ziagen) 960 mL per 30 days BG abacavir 300 mg tablet -

WHO-EMP-RHT-TSN-2018.1-Eng.Pdf
WHO/EMP/RHT/TSN/2018.1 The use of stems in the selection of International Nonproprietary Names (INN) for pharmaceutical substances FORMER DOCUMENT NUMBER: WHO/PHARM S/NOM 15 WHO/EMP/RHT/TSN/2018.1 © World Health Organization [2018] Some rights reserved. This work is available under the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 IGO licence (CC BY-NC-SA 3.0 IGO; https://creativecommons.org/licenses/by-nc-sa/3.0/igo). Under the terms of this licence, you may copy, redistribute and adapt the work for non-commercial purposes, provided the work is appropriately cited, as indicated below. In any use of this work, there should be no suggestion that WHO endorses any specific organization, products or services. The use of the WHO logo is not permitted. If you adapt the work, then you must license your work under the same or equivalent Creative Commons licence. If you create a translation of this work, you should add the following disclaimer along with the suggested citation: “This translation was not created by the World Health Organization (WHO). WHO is not responsible for the content or accuracy of this translation. The original English edition shall be the binding and authentic edition”. Any mediation relating to disputes arising under the licence shall be conducted in accordance with the mediation rules of the World Intellectual Property Organization. Suggested citation. The use of stems in the selection of International Nonproprietary Names (INN) for pharmaceutical substances. Geneva: World Health Organization; 2018 (WHO/EMP/RHT/TSN/2018.1). Licence: CC BY-NC-SA 3.0 IGO.