K. ENZYME KINETICS and CATALYSIS Basic Kinetics
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
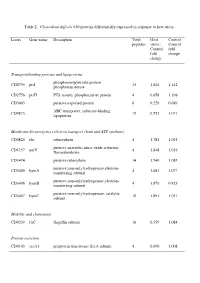
Table 2: Clostridium Difficile 630 Proteins Differentially Expressed in Response to Heat Stress
Table 2: Clostridium difficile 630 proteins differentially expressed in response to heat stress. Locus Gene name Description Total Heat Control : peptides stress : Control Control fold fold change change Transport/binding proteins and lipoproteins phosphoenolpyruvate-protein CD2755 ptsI 15 1.644 1.142 phosphotransferase CD2756 ptsH PTS system, phosphocarrier protein 4 0.658 1.198 CD3043 putative exported protein 6 0.229 0.689 ABC transporter, substrate-binding CD0873 15 0.521 1.071 lipoprotein Membrane bioenergetics (electron transport chain and ATP synthase) CD0825 rbr rubrerythrin 4 1.781 1.035 putative anaerobic nitric oxide reductase CD1157 norV 4 1.848 1.016 flavorubredoxin CD1474 putative ruberythrin 14 1.540 1.085 putative iron-only hydrogenase,electron- CD3405 hymA 4 1.681 1.077 transferring subunit putative iron-only hydrogenase,electron- CD3406 hymB 4 1.870 0.923 transferring subunit putative iron-only hydrogenase, catalytic CD3407 hymC 10 1.891 1.051 subunit Mobility and chemotaxis CD0239 fliC flagellin subunit 16 0.559 1.084 Protein secretion CD0143 secA1 preprotein translocase SecA subunit 4 0.690 1.008 Specific pathways putative oxidoreductase, thiamine diP-binding CD0116 13 1.628 1.121 subunit putative oxidoreductase, acetyl-CoA synthase CD0174 4 2.687 1.055 subunit CD0790 putative NUDIX-family hydrolase 8 1.491 1.32 inosine-uridine preferring nucleoside CD1682 iunH 4 0.518 0.984 hydrolase CD2181 putative aromatic compounds hydrolase 7 0372 0.909 CD2342 sucD succinate-semialdehyde dehydrogenase 6 0.651 0.906 Metabolism -

Generated by SRI International Pathway Tools Version 25.0, Authors S
Authors: Pallavi Subhraveti Ron Caspi Quang Ong Peter D Karp An online version of this diagram is available at BioCyc.org. Biosynthetic pathways are positioned in the left of the cytoplasm, degradative pathways on the right, and reactions not assigned to any pathway are in the far right of the cytoplasm. Transporters and membrane proteins are shown on the membrane. Ingrid Keseler Periplasmic (where appropriate) and extracellular reactions and proteins may also be shown. Pathways are colored according to their cellular function. Gcf_000725805Cyc: Streptomyces xanthophaeus Cellular Overview Connections between pathways are omitted for legibility. -

Bacterial Selenoproteins: a Role in Pathogenesis and Targets for Antimicrobial Development
University of Central Florida STARS Electronic Theses and Dissertations, 2004-2019 2009 Bacterial Selenoproteins: A Role In Pathogenesis And Targets For Antimicrobial Development Sarah Rosario University of Central Florida Part of the Medical Sciences Commons Find similar works at: https://stars.library.ucf.edu/etd University of Central Florida Libraries http://library.ucf.edu This Doctoral Dissertation (Open Access) is brought to you for free and open access by STARS. It has been accepted for inclusion in Electronic Theses and Dissertations, 2004-2019 by an authorized administrator of STARS. For more information, please contact [email protected]. STARS Citation Rosario, Sarah, "Bacterial Selenoproteins: A Role In Pathogenesis And Targets For Antimicrobial Development" (2009). Electronic Theses and Dissertations, 2004-2019. 3822. https://stars.library.ucf.edu/etd/3822 BACTERIAL SELENOPROTEINS: A ROLE IN PATHOGENESIS AND TARGETS FOR ANTIMICROBIAL DEVELOPMENT. by SARAH E. ROSARIO B.S. Florida State University, 2000 M.P.H. University of South Florida, 2002 A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Burnett School of Biomedical Sciences in the College of Medicine at the University of Central Florida Orlando, Florida Summer Term 2009 Major Professor: William T. Self © 2009 Sarah E. Rosario ii ABSTRACT Selenoproteins are unique proteins in which selenocysteine is inserted into the polypeptide chain by highly specialized translational machinery. They exist within all three kingdoms of life. The functions of these proteins in biology are still being defined. In particular, the importance of selenoproteins in pathogenic microorganisms has received little attention. We first established that a nosocomial pathogen, Clostridium difficile, utilizes a selenoenzyme dependent pathway for energy metabolism. -

Designed Mono- and Di-Covalent Inhibitors Trap Modeled Functional Motions for Trypanosoma Cruzi Proline Racemase in Crystallography
RESEARCH ARTICLE Designed mono- and di-covalent inhibitors trap modeled functional motions for Trypanosoma cruzi proline racemase in crystallography 1¤a 2¤b 2 Patricia de Aguiar Amaral , Delphine Autheman , Guilherme Dias de MeloID , 1 1 2¤c 2¤d Nicolas Gouault , Jean-FrancËois Cupif , Sophie GoyardID , Patricia Dutra , 2 2 3 4 4 Nicolas Coatnoan , Alain Cosson , Damien Monet , Frederick Saul , Ahmed HaouzID , 1 3 2¤e a1111111111 Philippe Uriac *, Arnaud Blondel *, Paola MinoprioID * a1111111111 a1111111111 1 Universite de Rennes 1, Equipe Chimie organique et interfaces (CORINT), UMR 6226 Sciences Chimiques de Rennes, Rennes, France, 2 Institut Pasteur, Laboratoire des Processus Infectieux à TrypanosomatideÂs, a1111111111 DeÂpartement Infection et EpideÂmiologie, Paris, France, 3 Institut Pasteur, Unite de Bioinformatique a1111111111 Structurale, DeÂpartement de Biologie Structurale et Chimie, CNRS-UMR 3528, Paris, France, 4 Institut Pasteur, Plateforme de Cristallographie, DeÂpartement de Biologie Structurale et Chimie, CNRS-UMR 3528, Paris, France ¤a Current address: LaPlaM/PPGCA, Universidade do Extremo Sul Catarinense, CriciuÂmaÐSC, Brazil ¤b Current address: Cell Surface Signalling Laboratory, Wellcome Trust Sanger Institute, Wellcome Trust OPEN ACCESS Genome Campus, Cambridge, United Kingdom Citation: Amaral PdA, Autheman D, de Melo GD, ¤c Current address: Institut Pasteur, Centre d'Innovation et Recherche Technologique, Paris, France Gouault N, Cupif J-F, Goyard S, et al. (2018) ¤d Current address: LaboratoÂrio de BioquõÂmica de ProtozoaÂrios e Imunofisiologia do ExercõÂcio, Departamento de Microbiologia, Imunologia e Parasitologia, Universidade do Estado do Rio de Janeiro, Rio Designed mono- and di-covalent inhibitors trap de Janeiro, Brazil modeled functional motions for Trypanosoma cruzi ¤e Current address: Trypanosomatids Infectious Processes Laboratory, Scientific Platform Pasteur-USP, proline racemase in crystallography. -

Discovery of a Novel Amino Acid Racemase Through Exploration of Natural Variation in Arabidopsis Thaliana
Discovery of a novel amino acid racemase through exploration of natural variation in Arabidopsis thaliana Renee C. Straucha,b, Elisabeth Svedinc, Brian Dilkesc, Clint Chappled, and Xu Lia,b,1 aPlants for Human Health Institute, North Carolina State University, Kannapolis, NC 28081; bDepartment of Plant and Microbial Biology, North Carolina State University, Raleigh, NC 27695; cDepartment of Horticulture and Landscape Architecture, Purdue University, West Lafayette, IN 47907; and dDepartment of Biochemistry, Purdue University, West Lafayette, IN 47907 Edited by Justin O. Borevitz, Australian National University, Canberra, ACT, Australia, and accepted by the Editorial Board August 1, 2015 (received for review February 16, 2015) Plants produce diverse low-molecular-weight compounds via spe- contribution to the variation in at least three-fourths of detected cialized metabolism. Discovery of the pathways underlying produc- mass peaks (8). tion of these metabolites is an important challenge for harnessing Here we describe an integrated transdisciplinary platform, the huge chemical diversity and catalytic potential in the plant king- combining metabolomics, genetics, and genomics, to exploit the dom for human uses, but this effort is often encumbered by the biochemical and genetic diversity of natural accessions of the necessity to initially identify compounds of interest or purify a cata- model plant A. thaliana to uncover associations between genes lyst involved in their synthesis. As an alternative approach, we have and metabolites. Using this platform, we linked a differentially performed untargeted metabolite profiling and genome-wide asso- accumulating metabolite, identified through chemical analysis as ciation analysis on 440 natural accessions of Arabidopsis thaliana. N-malonyl-D-allo-isoleucine (NMD-Ile), to a previously unchar- This approach allowed us to establish genetic linkages between acterized gene identified as an amino acid racemase through metabolites and genes. -

NIH Public Access Author Manuscript Curr Opin Chem Biol
NIH Public Access Author Manuscript Curr Opin Chem Biol. Author manuscript; available in PMC 2010 October 1. NIH-PA Author ManuscriptPublished NIH-PA Author Manuscript in final edited NIH-PA Author Manuscript form as: Curr Opin Chem Biol. 2009 October ; 13(4): 468±476. doi:10.1016/j.cbpa.2009.06.023. Pyridoxal 5'-Phosphate: Electrophilic Catalyst Extraordinaire John P. Richard†,*, Tina L. Amyes†, Juan Crugeiras#, and Ana Rios# †Department of Chemistry, University at Buffalo, SUNY, Buffalo, NY 14260-3000, USA. #Departamento de Química Física, Facultad de Química, Universidad de Santiago, 15782 Santiago de Compostela, Spain. Abstract Studies of nonenzymatic electrophilic catalysis of carbon deprotonation of glycine show that pyridoxal 5'-phosphate (PLP) strongly enhances the carbon acidity of α-amino acids, but that this is not the overriding mechanistic imperative for cofactor catalysis. Although the fully protonated PLP-glycine iminium ion adduct exhibits an extraordinary low α-imino carbon acidity (pKa = 6), the more weakly acidic zwitterionic iminium ion adduct (pKa = 17) is selected for use in enzymatic reactions. The similar α-imino carbon acidities of the iminium ion adducts of glycine with 5'- deoxypyridoxal and with phenylglyoxylate shows that the cofactor pyridine nitrogen plays a relatively minor role in carbanion stabilization. The 5'-phosphodianion group of PLP likely plays an important role in catalysis by providing up to 12 kcal/mol of binding energy that may be utilized for transition state stabilization. Introduction Scientists prize the rush of adrenalin that comes with making an original discovery, or with bringing order to seemingly disconnected experimental observations. Snell and Braunstein must have received tremendous satisfaction from their independent discovery, more than sixty years ago, that heating pyridoxal 5'-phosphate (PLP) with an amino acid yields the products of transamination of the amino acid [1]. -

SI Appendix Index 1
SI Appendix Index Calculating chemical attributes using EC-BLAST ................................................................................ 2 Chemical attributes in isomerase reactions ............................................................................................ 3 Bond changes …..................................................................................................................................... 3 Reaction centres …................................................................................................................................. 5 Substrates and products …..................................................................................................................... 6 Comparative analysis …........................................................................................................................ 7 Racemases and epimerases (EC 5.1) ….................................................................................................. 7 Intramolecular oxidoreductases (EC 5.3) …........................................................................................... 8 Intramolecular transferases (EC 5.4) ….................................................................................................. 9 Supporting references …....................................................................................................................... 10 Fig. S1. Overview …............................................................................................................................ -

Proline Racemases: Insights Into Trypanosoma Cruzi Peptides Containing D-Proline
Mem Inst Oswaldo Cruz, Rio de Janeiro, Vol. 104(Suppl. I): 295-300, 2009 295 Proline racemases: insights into Trypanosoma cruzi peptides containing D-proline Nicolas Coatnoan, Armand Berneman, Nathalie Chamond, Paola Minoprio/+ Institut Pasteur, Laboratoire d’Immunobiologie des Infections à Trypanosoma, Département d’Immunologie, 25 Rue du Dr. Roux, 75724 Paris, Cedex 15, France Trypanosoma cruzi proline racemases (TcPRAC) are homodimeric enzymes that interconvert the L and D- enantiomers of proline. At least two paralogous copies of proline racemase (PR) genes are present per parasite haploid genome and they are differentially expressed during T. cruzi development. Non-infective epimastigote forms that overexpress PR genes differentiate more readily into metacyclic infective forms that are more invasive to host cells, indicating that PR participates in mechanisms of virulence acquisition. Using a combination of biochemi- cal and enzymatic methods, we show here that, in addition to free D-amino acids, non-infective epimastigote and infective metacyclic parasite extracts possess peptides composed notably of D-proline. The relative contribution of TcPRAC to D-proline availability and its further assembly into peptides was estimated through the use of wild-type parasites and parasites over-expressing TcPRAC genes. Our data suggest that D-proline-bearing peptides, similarly to the mucopeptide layer of bacterial cell walls, may be of benefit to T. cruzi by providing resistance against host proteolytic mechanisms. Key words: proline racemase - TcPRAC - D-proline The role of D-amino acids (D-AA) in physiopatho- D-AAs to provide an envelope composed of peptidogly- logical processes has occupied the attention of several cans, teichoic acid, polysaccharides and proteins (Mar- groups for many years. -

Crystal Structures of Streptomyces Coelicolor Methylmalonyl-Coa Epi
Crystal structures of Streptomyces coelicolor methylmalonyl-CoA epi- merase with substrate or transition state analog contradicts a simple general acid-base catalytic mechanism Lee M. Stunkard, Aaron B. Benjamin, James B. Bower, Tyler J. Huth and Jeremy R. Lohman* Department of Biochemistry, Purdue University Center for Cancer Research, Purdue University, West Lafayette, Indiana 47907, United States Supporting Information Placeholder ABSTRACT: Isomerases that flip carbon stereocenter chirality intermediary metabolism, but also for the production of polyketides often use straightforward general acid base catalysis where a proton in certain heterologous hosts. is abstracted from one face of a central carbon and a proton is de- Another source of methylmalonyl-CoA for polyketide biosyn- posited on the opposite. That mechanism has been ascribed to thesis is through the activity of malonyl-CoA synthetases such as methylmalonyl-CoA epimerase (MMCE). The proposed mecha- MatB from S. coelicolor or Rhizobium trifolii although they gener- nism is use of concerted general acid-base catalysis similar to the ate (2R)-methylmalonyl-CoA thus also requiring MMCE to sup- proline racemase, based on the exchange of the α-carbon proton port efficient DEBS-like polyketide biosynthesis.5-6 MatB enzymes with solvent. Tritium kinetic isotope effect experiments lead to the also accept substrates other than malonate and methylmalonate hypothesis of a “protected base” in catalysis. Structures of Propi- with poor efficiency,7 although mutants have been generated with onibacterium shermanii MMCE with catalytic Co2+ bound revealed expanded substrate specificity.8 Therefore, an attractive path to that two glutamates were positioned on opposite sides of the active polyketide derivatives is through the engineering of MatB to pro- site, that could fulfil the roles of the general acids. -
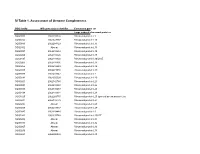
SI Table 1. Assessment of Genome Completeness
SI Table 1. Assessment of Genome Completeness COG family IMG gene object identifier Conserved gene set Large subunit ribosomal proteins COG0081 2062288324 Ribosomal protein L1 COG0244 2062347387 Ribosomal protein L10 COG0080 2062288323 Ribosomal protein L11 COG0102 Absent Ribosomal protein L13 COG0093 2062418832 Ribosomal protein L14 COG0200 2062418826 Ribosomal protein L15 COG0197 2062418838 Ribosomal protein L16/L10E COG0203 2062418836 Ribosomal protein L17 COG0256 2062418829 Ribosomal protein L18 COG0335 2062273558 Ribosomal protein L19 COG0090 2062418842 Ribosomal protein L2 COG0292 2062350539 Ribosomal protein L20 COG0261 2062142780 Ribosomal protein L21 COG0091 2062418840 Ribosomal protein L22 COG0089 2062138283 Ribosomal protein L23 COG0198 2062418834 Ribosomal protein L24 COG1825 2062269715 Ribosomal protein L25 (general stress protein Ctc) COG0211 2062142779 Ribosomal protein L27 COG0227 Absent Ribosomal protein L28 COG0255 2062418837 Ribosomal protein L29 COG0087 2062154483 Ribosomal protein L3 COG1841 2062335748 Ribosomal protein L30/L7E COG0254 Absent Ribosomal protein L31 COG0333 Absent Ribosomal protein L32 COG0267 Absent Ribosomal protein L33 COG0230 Absent Ribosomal protein L34 COG0291 2062350538 Ribosomal protein L35 COG0257 Absent Ribosomal protein L36 COG0088 2062138282 Ribosomal protein L4 COG0094 2062418833 Ribosomal protein L5 COG0097 2062418830 Ribosomal protein L6P/L9E COG0222 2062288326 Ribosomal protein L7/L12 COG0359 2062209880 Ribosomal protein L9 Small subunit ribosomal proteins COG0539 Absent Ribosomal protein -

Drug Discovery Targeting Amino Acid Racemases Paola Conti, Lucia Tamborini, Andrea Pinto, Arnaud Blondel, Paola Minoprio, Andrea Mozzarelli, Carlo De Micheli
Drug Discovery Targeting Amino Acid Racemases Paola Conti, Lucia Tamborini, Andrea Pinto, Arnaud Blondel, Paola Minoprio, Andrea Mozzarelli, Carlo de Micheli To cite this version: Paola Conti, Lucia Tamborini, Andrea Pinto, Arnaud Blondel, Paola Minoprio, et al.. Drug Discovery Targeting Amino Acid Racemases. Chemical Reviews, American Chemical Society, 2011, 111 (11), pp.6919-6946. 10.1021/cr2000702. pasteur-02510858 HAL Id: pasteur-02510858 https://hal-pasteur.archives-ouvertes.fr/pasteur-02510858 Submitted on 7 Apr 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Drug Discovery Targeting Amino Acid Racemases Paola Conti,a Lucia Tamborini, a Andrea Pinto,a Arnaud Blondel,b Paola Minoprio,c Andrea Mozzarellid and Carlo De Micheli a* aDipartimento di Scienze Farmaceutiche ‘P. Pratesi’, via Mangiagalli 25, 20133 Milano, Italy; bUnité de Bioinformatique Structurale, CNRS-URA 2185, Département de Biologie Structurale et Chimie, 25 rue du Dr. Roux, 75724 Paris, France. cInstitut Pasteur, Laboratoire des Processus Infectieux à Trypanosoma; Départment d’Infection and Epidémiologie; 25 rue du Dr. Roux, 75724 Paris, France dDipartimento di Biochimica e Biologia Molecolare, via G. P. Usberti 23/A, 43100 Parma, Italy and Istituto di Biostrutture e Biosistemi, viale Medaglie d’oro, Rome, Italy. -

Substrate-Induced Conformational Changes in Bacillus Subtilis Glutamate Racemase and Their Implications for Drug Discovery
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Structure, Vol. 13, 1707–1713, November, 2005, ª2005 Elsevier Ltd All rights reserved. DOI 10.1016/j.str.2005.07.024 Substrate-Induced Conformational Changes in Bacillus subtilis Glutamate Racemase and Their Implications for Drug Discovery Sergey N. Ruzheinikov,1 Makie A. Taal,1 that operate in a cofactor-independent fashion and that Svetlana E. Sedelnikova, Patrick J. Baker, includes proline racemase, aspartate racemase, and di- and David W. Rice* aminopimelate epimerase. In Streptococcus pneumo- Krebs Institute for Biomolecular Research niae (Baltz et al., 1999), Escherichia coli (Doublet et al., Department of Molecular Biology and Biotechnology 1992, 1993), Staphylococcus haemolyticus (Pucci et al., University of Sheffield 1995), and Bacillus subtilis (Kobayashi et al., 2003), the Firth Court presence of a functional RacE has been shown to be es- Western Bank sential for cell viability and suggests that this enzyme Sheffield S10 2TN forms a possible target for the development of antibacte- United Kingdom rial agents. Moreover, since D-Glu is not normally found in mammalian hosts, drugs based on a D-Glu framework might be expected to have low toxicity (Bugg and Walsh, Summary 1992; Walsh, 1989). Recently the first potent inhibitors of RacE, a series of D-substituted amino acids, showing D-glutamate is an essential building block of the pep- whole-cell antibacterial activity, have been described tidoglycan layer in bacterial cell walls and can be syn- (de Dios et al., 2002). thesized from L-glutamate by glutamate racemase The structure of Aquifex pyrophilus RacE provided the (RacE).