Bacterial Selenoproteins: a Role in Pathogenesis and Targets for Antimicrobial Development
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Effect of Bath Temperature on the Electrodeposition of Snse Thin
The 4th Annual Seminar of National Science Fellowship 2004 [AMT10] Aqueous electrodeposition and properties of tin selenide thin films Saravanan Nagalingam1, Zulkarnain Zainal1, Anuar Kassim1, Mohd. Zobir Hussein1, Wan Mahmood Mat Yunus2 1Department of Chemistry, 2Department of Physics, Faculty of Science, Universiti Putra Malaysia, 43400 Serdang, Selangor, Malaysia Introduction Motivated by the potential applications of composition of the electrolytic solution tin chalcogenides, investigations on these (Riveros et al., 2001). We report here the compounds are becoming particularly active electrodeposition of SnSe thin films under in the field of materials chemistry. Tin aqueous conditions in the presence of chalcogenides offer a range of optical band ethylendiaminetetraacetate (EDTA) as a gaps suitable for various optical and chelating agent. optoelectronic applications. These compounds are also used as sensor and laser materials, Materials and Methods thin films polarizers and thermoelectric A conventional three-electrode cell was cooling materials (Zweibel, 2000). employed in this study. Ag/AgCl was used as Considerable attention has been given by the reference electrode to which all potentials various researchers in studying the properties were quoted. The working and counter of tin selenide (SnSe). SnSe is a narrow band electrodes were made from indium tin oxide gap binary IV-VI semiconductor with an (ITO) glass substrate and platinum, orthorhombic crystal structure. Among the respectively. The ITO glass substrates were uses of tin selenide (SnSe) are as memory cleaned ultrasonically in ethanol and distilled switching devices, holographic recording water before the deposition process. The systems, and infrared electronic devices counter electrode was polished prior to the (Lindgren et al., 2002). SnSe has been studied insertion into the electrolyte cell. -
![Electronic Structure of Designed [(Snse)1+D ]M [Tise2]](https://docslib.b-cdn.net/cover/8052/electronic-structure-of-designed-snse-1-d-m-tise2-98052.webp)
Electronic Structure of Designed [(Snse)1+D ]M [Tise2]
Invited Paper DOI: 10.1557/jmr.2019.128 Electronic structure of designed [(SnSe)1+d]m[TiSe2]2 heterostructure thin films with tunable layering sequence https://doi.org/10.1557/jmr.2019.128 . Fabian Göhler1 , Danielle M. Hamann2, Niels Rösch1, Susanne Wolff1, Jacob T. Logan2, Robert Fischer2, Florian Speck1, David C. Johnson2, Thomas Seyller1,a) 1Institute of Physics, Chemnitz University of Technology, D-09126 Chemnitz, Germany 2Department of Chemistry, University of Oregon, Eugene, Oregon 97401, USA a)Address all correspondence to this author. e-mail: [email protected] Received: 18 February 2019; accepted: 21 March 2019 fi m A series of [(SnSe)1+d]m[TiSe2]2 heterostructure thin lms built up from repeating units of bilayers of SnSe and two layers of TiSe2 were synthesized from designed precursors. The electronic structure of the films was https://www.cambridge.org/core/terms investigated using X-ray photoelectron spectroscopy for samples with m = 1, 2, 3, and 7 and compared to binary samples of TiSe2 and SnSe. The observed binding energies of core levels and valence bands of the heterostructures are largely independent of m. For the SnSe layers, we can observe a rigid band shift in the heterostructures compared to the binary, which can be explained by electron transfer from SnSe to TiSe2. The electronic structure of the TiSe2 layers shows a more complicated behavior, as a small shift can be observed in the valence band and Se3d spectra, but the Ti2p core level remains at a constant energy. Complementary UV photoemission spectroscopy measurements confirm a charge transfer mechanism where the SnSe layers donate electrons into empty Ti3d states at the Fermi energy. -

Amino Acid Catabolism in Staphylococcus Aureus
University of Nebraska Medical Center DigitalCommons@UNMC Theses & Dissertations Graduate Studies Fall 12-16-2016 Amino Acid Catabolism in Staphylococcus aureus Cortney Halsey University of Nebraska Medical Center Follow this and additional works at: https://digitalcommons.unmc.edu/etd Part of the Bacteriology Commons Recommended Citation Halsey, Cortney, "Amino Acid Catabolism in Staphylococcus aureus" (2016). Theses & Dissertations. 160. https://digitalcommons.unmc.edu/etd/160 This Dissertation is brought to you for free and open access by the Graduate Studies at DigitalCommons@UNMC. It has been accepted for inclusion in Theses & Dissertations by an authorized administrator of DigitalCommons@UNMC. For more information, please contact [email protected]. Amino Acid Catabolism in Staphylococcus aureus By Cortney R. Halsey A DISSERTATION Presented to the Faculty of The Graduate College in the University of Nebraska In Partial Fulfillment of the Requirements For the Degree of Doctor of Philosophy Pathology and Microbiology Under the Supervision of Dr. Paul D. Fey University of Nebraska Medical Center Omaha, Nebraska October 2016 Supervisory Committee: Kenneth Bayles, Ph.D. Steven Carson, Ph.D. Paul Dunman, Ph.D. Rakesh Singh, Ph.D. ii Acknowledgements First and foremost, I would like to thank my mentor, Dr. Paul Fey, whose patience and support over the past six years has been critical to my success as a graduate student. Paul has given me opportunities to grow as a scientist and person, for which I will be forever thankful. I would also like to thank Dr. Ken Bayles, Dr. Steven Carson, Dr. Paul Dunman, and Dr. Rakesh Singh for serving on my supervisory committee. -

New Insights Into Clostridia Through Comparative Analyses of Their 40 Genomes
Bioenerg. Res. DOI 10.1007/s12155-014-9486-9 New Insights into Clostridia Through Comparative Analyses of Their 40 Genomes Chuan Zhou & Qin Ma & Xizeng Mao & Bingqiang Liu & Yanbin Yin & Ying Xu # Springer Science+Business Media New York 2014 Abstract The Clostridium genus of bacteria contains the gene groups are shared by all the strains, and the strain- most widely studied biofuel-producing organisms such as specific gene pool consists of 22,668 genes, which is consis- Clostridium thermocellum and also some human pathogens, tent with the fact that these bacteria live in very diverse plus a few less characterized strains. Here, we present a environments and have evolved a very large number of comparative genomic analysis of 40 fully sequenced clostrid- strain-specific genes to adapt to different environments. ial genomes, paying a particular attention to the biomass Across the 40 genomes, 1.4–5.8 % of genes fall into the degradation ones. Our analysis indicates that some of the carbohydrate active enzyme (CAZyme) families, and 20 out Clostridium botulinum strains may have been incorrectly clas- of the 40 genomes may encode cellulosomes with each ge- sified in the current taxonomy and hence should be renamed nome having 1 to 76 genes bearing the cellulosome-related according to the 16S ribosomal RNA (rRNA) phylogeny. A modules such as dockerins and cohesins. A phylogenetic core-genome analysis suggests that only 169 orthologous footprinting analysis identified cis-regulatory motifs that are enriched in the promoters of the CAZyme genes, giving rise to 32 statistically significant motif candidates. Keywords Clostridium . Comparative genomics . -
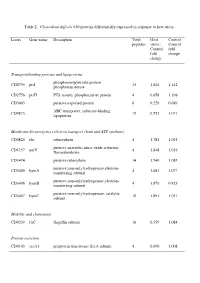
Table 2: Clostridium Difficile 630 Proteins Differentially Expressed in Response to Heat Stress
Table 2: Clostridium difficile 630 proteins differentially expressed in response to heat stress. Locus Gene name Description Total Heat Control : peptides stress : Control Control fold fold change change Transport/binding proteins and lipoproteins phosphoenolpyruvate-protein CD2755 ptsI 15 1.644 1.142 phosphotransferase CD2756 ptsH PTS system, phosphocarrier protein 4 0.658 1.198 CD3043 putative exported protein 6 0.229 0.689 ABC transporter, substrate-binding CD0873 15 0.521 1.071 lipoprotein Membrane bioenergetics (electron transport chain and ATP synthase) CD0825 rbr rubrerythrin 4 1.781 1.035 putative anaerobic nitric oxide reductase CD1157 norV 4 1.848 1.016 flavorubredoxin CD1474 putative ruberythrin 14 1.540 1.085 putative iron-only hydrogenase,electron- CD3405 hymA 4 1.681 1.077 transferring subunit putative iron-only hydrogenase,electron- CD3406 hymB 4 1.870 0.923 transferring subunit putative iron-only hydrogenase, catalytic CD3407 hymC 10 1.891 1.051 subunit Mobility and chemotaxis CD0239 fliC flagellin subunit 16 0.559 1.084 Protein secretion CD0143 secA1 preprotein translocase SecA subunit 4 0.690 1.008 Specific pathways putative oxidoreductase, thiamine diP-binding CD0116 13 1.628 1.121 subunit putative oxidoreductase, acetyl-CoA synthase CD0174 4 2.687 1.055 subunit CD0790 putative NUDIX-family hydrolase 8 1.491 1.32 inosine-uridine preferring nucleoside CD1682 iunH 4 0.518 0.984 hydrolase CD2181 putative aromatic compounds hydrolase 7 0372 0.909 CD2342 sucD succinate-semialdehyde dehydrogenase 6 0.651 0.906 Metabolism -

PRAJNA - Journal of Pure and Applied Sciences ISSN 0975 2595 Volume 19 December 2011 CONTENTS
PRAJNA - Journal of Pure and Applied Sciences ISSN 0975 2595 Volume 19 December 2011 CONTENTS BIOSCIENCES Altered energy transfer in Phycobilisomes of the Cyanobacterium, Spirulina Platensis under 1 - 3 the influence of Chromium (III) Ayya Raju, M. and Murthy, S. D. S. PRAJNA Volume 19, 2011 Biotransformation of 11β , 17 α -dihydroxy-4-pregnene-3, 20-dione-21-o-succinate to a 4 - 7 17-ketosteroid by Pseudomonas Putida MTCC 1259 in absence of 9α -hydroxylase inhibitors Rahul Patel and Kirti Pawar Influence of nicking in combination with various plant growth substances on seed 8 - 10 germination and seedling growth of Noni (Morinda Citrifolia L.) Karnam Jaya Chandra and Dasari Daniel Gnana Sagar Quantitative analysis of aquatic Macrophytes in certain wetlands of Kachchh District, 11 - 13 Journal of Pure and Applied Sciences Gujarat J.P. Shah, Y.B. Dabgar and B.K. Jain Screening of crude root extracts of some Indian plants for their antibacterial activity 14 - 18 Purvesh B. Bharvad, Ashish R. Nayak, Naynika K. Patel and J. S. S. Mohan ________ Short Communication Heterosis for biometric characters and seed yield in parents and hybrids of rice 19 - 20 (Oryza Sativa L.) M. Prakash and B. Sunil Kumar CHEMISTRY Adsorption behavior and thermodynamics investigation of Aniline-n- 21 - 24 (p-Methoxybenzylidene) as corrosion inhibitor for Al-Mg alloy in hydrochloric acid V.A. Panchal, A.S. Patel and N.K. Shah Grafting of Butyl Acrylate onto Sodium Salt of partially Carboxymethylated Guar Gum 25 - 31 using Ceric Ions J.H. Trivedi, T.A. Bhatt and H.C. Trivedi Simultaneous equation and absorbance ratio methods for estimation of Fluoxetine 32 - 36 Hydrochloride and Olanzapine in tablet dosage form Vijaykumar K. -

Gastroenterology
Gastroenterology Inhibiting Growth of Clostridioides difficile by Restoring Valerate, Produced by the Intestinal Microbiota --Manuscript Draft-- Manuscript Number: GASTRO-D-18-00449R1 Full Title: Inhibiting Growth of Clostridioides difficile by Restoring Valerate, Produced by the Intestinal Microbiota Article Type: Basic - Alimentary Tract Corresponding Author: Julian Marchesi Cardiff University Cardiff, UNITED KINGDOM Corresponding Author Secondary Information: Corresponding Author's Institution: Cardiff University Corresponding Author's Secondary Institution: First Author: Julie A. K. McDonald, PhD First Author Secondary Information: Order of Authors: Julie A. K. McDonald, PhD Benjamin H. Mullish, MB BChir Alexandros Pechlivanis, PhD Zhigang Liu, PhD Jerusa Brignardello, MSc Dina Kao, MD Elaine Holmes, PhD Jia V. Li, PhD Thomas B Clarke, PhD Mark R. Thursz, MD Julian R. Marchesi, PhD Order of Authors Secondary Information: Abstract: Background & Aims: Faecal microbiota transplantation (FMT) is effective for treating recurrent Clostridioides difficile infection (CDI), but there are concerns regarding its long-term safety. Understanding the mechanisms of the effects of FMT could help us design safer, targeted therapies. We aimed to identify microbial metabolites that are important for C difficile growth. Methods: We used a CDI chemostat model as a tool to study the effects of FMT in vitro. The following analyses were performed: C difficile plate counts, 16S rRNA gene sequencing, 1H-NMR spectroscopy, and UPLC mass spectrometry bile acid profiling. FMT mixtures were prepared using fresh faecal samples provided by donors enrolled in the FMT Programme. Results from chemostat experiments were validated using human stool samples, C difficile batch cultures, and C57BL/6 mice with CDI. Human stool samples were collected from 16 patients with recurrent CDI and 5 healthy donors participating in an FMT trial. -

Designing Isoelectronic Counterparts to Layered Group V Semiconductors
Designing isoelectronic counterparts to layered group V semiconductors Zhen Zhu, Jie Guan, Dan Liu, and David Tom´anek∗ Physics and Astronomy Department, Michigan State University, East Lansing, Michigan 48824, USA In analogy to III-V compounds, which have significantly broadened the scope of group IV semi- conductors, we propose IV-VI compounds as isoelectronic counterparts to layered group V semi- conductors. Using ab initio density functional theory, we study yet unrealized structural phases of silicon mono-sulfide (SiS). We find the black-phosphorus-like α-SiS to be almost equally stable as the blue-phosphorus-like β-SiS. Both α-SiS and β-SiS monolayers display a significant, indirect band gap that depends sensitively on the in-layer strain. Unlike 2D semiconductors of group V elements with the corresponding nonplanar structure, different SiS allotropes show a strong polarization either within or normal to the layers. We find that SiS may form both lateral and vertical heterostructures with phosphorene at a very small energy penalty, offering an unprecedented tunability in structural and electronic properties of SiS-P compounds. INTRODUCTION RESULTS AND DISCUSSION 2D semiconductors of group V elements, including Since all atoms in sp3 layered structures of group V el- phosphorene and arsenene, have been rapidly attract- ements are threefold coordinated, the different allotropes ing interest due to their significant fundamental band can all be topologically mapped onto the honeycomb lat- gap, large density of states near the Fermi level, and tice of graphene with 2 sites per unit cell.[11] An easy high and anisotropic carrier mobility[1{4]. Combina- way to generate IV-VI compounds that are isoelectronic tion of these properties places these systems very favor- to group V monolayers is to occupy one of these sites by ably in the group of contenders for 2D electronics ap- a group IV and the other by a group VI element. -

A Survey of Carbon Fixation Pathways Through a Quantitative Lens
Journal of Experimental Botany, Vol. 63, No. 6, pp. 2325–2342, 2012 doi:10.1093/jxb/err417 Advance Access publication 26 December, 2011 REVIEW PAPER A survey of carbon fixation pathways through a quantitative lens Arren Bar-Even, Elad Noor and Ron Milo* Department of Plant Sciences, The Weizmann Institute of Science, Rehovot 76100, Israel * To whom correspondence should be addressed. E-mail: [email protected] Received 15 August 2011; Revised 4 November 2011; Accepted 8 November 2011 Downloaded from Abstract While the reductive pentose phosphate cycle is responsible for the fixation of most of the carbon in the biosphere, it http://jxb.oxfordjournals.org/ has several natural substitutes. In fact, due to the characterization of three new carbon fixation pathways in the last decade, the diversity of known metabolic solutions for autotrophic growth has doubled. In this review, the different pathways are analysed and compared according to various criteria, trying to connect each of the different metabolic alternatives to suitable environments or metabolic goals. The different roles of carbon fixation are discussed; in addition to sustaining autotrophic growth it can also be used for energy conservation and as an electron sink for the recycling of reduced electron carriers. Our main focus in this review is on thermodynamic and kinetic aspects, including thermodynamically challenging reactions, the ATP requirement of each pathway, energetic constraints on carbon fixation, and factors that are expected to limit the rate of the pathways. Finally, possible metabolic structures at Weizmann Institute of Science on July 3, 2016 of yet unknown carbon fixation pathways are suggested and discussed. -

Generated by SRI International Pathway Tools Version 25.0, Authors S
Authors: Pallavi Subhraveti Ron Caspi Quang Ong Peter D Karp An online version of this diagram is available at BioCyc.org. Biosynthetic pathways are positioned in the left of the cytoplasm, degradative pathways on the right, and reactions not assigned to any pathway are in the far right of the cytoplasm. Transporters and membrane proteins are shown on the membrane. Ingrid Keseler Periplasmic (where appropriate) and extracellular reactions and proteins may also be shown. Pathways are colored according to their cellular function. Gcf_000725805Cyc: Streptomyces xanthophaeus Cellular Overview Connections between pathways are omitted for legibility. -

Chemistry of Proteins and Amino Acids • Proteins Are the Most Abundant Organic Molecules of the Living System
Chemistry of Proteins and Amino Acids • Proteins are the most abundant organic molecules of the living system. • They occur in the every part of the cell and constitute about 50% of the cellular dry weight. • Proteins form the fundamental basis of structure and function of life. • In 1839 Dutch chemist G.J.Mulder while investing the substances such as those found in milk, egg, found that they could be coagulated on heating and were nitrogenous compounds. • The term protein is derived from a Greek word proteios, meaning first place. • Berzelius ( Swedish chemist ) suggested the name proteins to the group of organic compounds that are utmost important to life. • The proteins are nitrogenous macromolecules composed of many amino acids. Biomedical importance of proteins: • Proteins are the main structural components of the cytoskeleton. They are the sole source to replace nitrogen of the body. • Bio chemical catalysts known as enzymes are proteins. • Proteins known as immunoglobulins serve as the first line of defense against bacterial and viral infections. • Several hormones are protein in nature. • Structural proteins like actin and myosin are contractile proteins and help in the movement of muscle fibre. Some proteins present in cell membrane, cytoplasm and nucleus of the cell act as receptors. • The transport proteins carry out the function of transporting specific substances either across the membrane or in the body fluids. • Storage proteins bind with specific substances and store them, e.g. iron is stored as ferritin. • Few proteins are constituents of respiratory pigments and occur in electron transport chain, e.g. Cytochromes, hemoglobin, myoglobin • Under certain conditions proteins can be catabolized to supply energy. -

EXPERIMENTAL STUDIES on FERMENTATIVE FIRMICUTES from ANOXIC ENVIRONMENTS: ISOLATION, EVOLUTION, and THEIR GEOCHEMICAL IMPACTS By
EXPERIMENTAL STUDIES ON FERMENTATIVE FIRMICUTES FROM ANOXIC ENVIRONMENTS: ISOLATION, EVOLUTION, AND THEIR GEOCHEMICAL IMPACTS By JESSICA KEE EUN CHOI A dissertation submitted to the School of Graduate Studies Rutgers, The State University of New Jersey In partial fulfillment of the requirements For the degree of Doctor of Philosophy Graduate Program in Microbial Biology Written under the direction of Nathan Yee And approved by _______________________________________________________ _______________________________________________________ _______________________________________________________ _______________________________________________________ New Brunswick, New Jersey October 2017 ABSTRACT OF THE DISSERTATION Experimental studies on fermentative Firmicutes from anoxic environments: isolation, evolution and their geochemical impacts by JESSICA KEE EUN CHOI Dissertation director: Nathan Yee Fermentative microorganisms from the bacterial phylum Firmicutes are quite ubiquitous in subsurface environments and play an important biogeochemical role. For instance, fermenters have the ability to take complex molecules and break them into simpler compounds that serve as growth substrates for other organisms. The research presented here focuses on two groups of fermentative Firmicutes, one from the genus Clostridium and the other from the class Negativicutes. Clostridium species are well-known fermenters. Laboratory studies done so far have also displayed the capability to reduce Fe(III), yet the mechanism of this activity has not been investigated