Substrate-Induced Conformational Changes in Bacillus Subtilis Glutamate Racemase and Their Implications for Drug Discovery
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
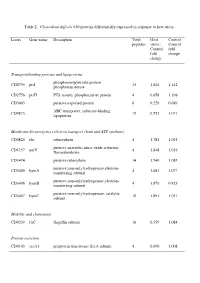
Table 2: Clostridium Difficile 630 Proteins Differentially Expressed in Response to Heat Stress
Table 2: Clostridium difficile 630 proteins differentially expressed in response to heat stress. Locus Gene name Description Total Heat Control : peptides stress : Control Control fold fold change change Transport/binding proteins and lipoproteins phosphoenolpyruvate-protein CD2755 ptsI 15 1.644 1.142 phosphotransferase CD2756 ptsH PTS system, phosphocarrier protein 4 0.658 1.198 CD3043 putative exported protein 6 0.229 0.689 ABC transporter, substrate-binding CD0873 15 0.521 1.071 lipoprotein Membrane bioenergetics (electron transport chain and ATP synthase) CD0825 rbr rubrerythrin 4 1.781 1.035 putative anaerobic nitric oxide reductase CD1157 norV 4 1.848 1.016 flavorubredoxin CD1474 putative ruberythrin 14 1.540 1.085 putative iron-only hydrogenase,electron- CD3405 hymA 4 1.681 1.077 transferring subunit putative iron-only hydrogenase,electron- CD3406 hymB 4 1.870 0.923 transferring subunit putative iron-only hydrogenase, catalytic CD3407 hymC 10 1.891 1.051 subunit Mobility and chemotaxis CD0239 fliC flagellin subunit 16 0.559 1.084 Protein secretion CD0143 secA1 preprotein translocase SecA subunit 4 0.690 1.008 Specific pathways putative oxidoreductase, thiamine diP-binding CD0116 13 1.628 1.121 subunit putative oxidoreductase, acetyl-CoA synthase CD0174 4 2.687 1.055 subunit CD0790 putative NUDIX-family hydrolase 8 1.491 1.32 inosine-uridine preferring nucleoside CD1682 iunH 4 0.518 0.984 hydrolase CD2181 putative aromatic compounds hydrolase 7 0372 0.909 CD2342 sucD succinate-semialdehyde dehydrogenase 6 0.651 0.906 Metabolism -

Fulltext01.Pdf
http://www.diva-portal.org This is the published version of a paper published in Cellular and Molecular Life Sciences (CMLS). Citation for the original published paper (version of record): Cava, F., Lam, H., de Pedro, M., Waldor, M. (2011) Emerging knowledge of regulatory roles of D-amino acids in bacteria. Cellular and Molecular Life Sciences (CMLS), 68(5): 817-831 http://dx.doi.org/10.1007/s00018-010-0571-8 Access to the published version may require subscription. N.B. When citing this work, cite the original published paper. Permanent link to this version: http://urn.kb.se/resolve?urn=urn:nbn:se:umu:diva-81861 Cell. Mol. Life Sci. (2011) 68:817–831 DOI 10.1007/s00018-010-0571-8 Cellular and Molecular Life Sciences REVIEW Emerging knowledge of regulatory roles of D-amino acids in bacteria Felipe Cava • Hubert Lam • Miguel A. de Pedro • Matthew K. Waldor Received: 13 July 2010 / Revised: 24 September 2010 / Accepted: 14 October 2010 / Published online: 14 December 2010 Ó The Author(s) 2010. This article is published with open access at Springerlink.com Abstract The D-enantiomers of amino acids have been Keywords D-amino acid Á Racemase Á Stationary phase Á thought to have relatively minor functions in biological Peptidoglycan Á Biofilm Á Regulation processes. While L-amino acids clearly predominate in nat- ure, D-amino acids are sometimes found in proteins that are Abbreviations not synthesized by ribosomes, and D-Ala and D-Glu are NRP Nonribosomal peptide routinely found in the peptidoglycan cell wall of bacteria. PG Peptidoglycan Here, we review recent findings showing that D-amino acids GlcNAc N-acetyl glucosamine have previously unappreciated regulatory roles in the bac- MurNAc N-acetylmuramic acid terial kingdom. -

Coordinated Slowing of Metabolism in Enteric Bacteria Under Nitrogen
Coordinated Slowing of Metabolism in Enteric Bacteria under Nitrogen Limitation: A Perspective Ned S. Wingreen NEC Research Institute, 4 Independence Way Princeton, New Jersey 08540 and Department of Physics, University of California Berkeley, CA 94720 Sydney Kustu Department of Plant Biology, Molecular and Cell Biology University of California, Berkeley, CA 94720 Abstract It is natural to ask how bacteria coordinate metabolism when depletion of an essential nutrient limits their growth, and they must slow their entire rate of biosyn- thesis. A major nutrient with a fluctuating abundance is nitrogen. The growth rate of enteric bacteria under nitrogen-limiting conditions is known to correlate with the internal concentration of free glutamine, the glutamine pool. Here we compare the patterns of utilization of L-glutamine and L-glutamate, the two central inter- mediates of nitrogen metabolism. Monomeric precursors of all of the cell’s macro- molecules – proteins, nucleic acids, and surface polymers – require the amide group of glutamine at the first dedicated step of biosynthesis. This is the case even though only a minority (∼12%) of total cell nitrogen derives from glutamine. In contrast, the amino group of glutamate, which provides the remainder of cell nitrogen, is arXiv:physics/0110037v1 [physics.bio-ph] 12 Oct 2001 generally required late in biosynthetic pathways, e.g. in transaminase reactions for amino acid synthesis. We propose that the pattern of glutamine dependence coor- dinates the decrease in biosynthesis under conditions of nitrogen limitation. Hence, the glutamine pool plays a global regulatory role in the cell. 1 INTRODUCTION Enteric bacteria are notable for their varying environment. -

Title Non-Stereospecific Transamination Catalyzed by Pyridoxal Phosphate-Dependent Amino Acid Racemases of Broad Substrate Speci
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Kyoto University Research Information Repository Non-stereospecific Transamination Catalyzed by Pyridoxal Phosphate-dependent Amino Acid Racemases of Broad Title Substrate Specificity (MOLECULAR BIOFUNCTION- Molecular Microbial Science) Esaki, Nobuyoshi; Yoshimura, Tohru; Soda, Kenji; Lim, Author(s) Young Hee Citation ICR annual report (1999), 5: 46-47 Issue Date 1999-03 URL http://hdl.handle.net/2433/65185 Right Type Article Textversion publisher Kyoto University 46 ICR Annual Report, Vol. 5, 1998 Non-stereospecific Transamination Catalyzed by Pyridoxal Phosphate-dependent Amino Acid Racemases of Broad Substrate Specificity Nobuyoshi Esaki, Tohru Yoshimura, Kenji Soda and Young Hee Lim Pyridoxal 5’-phosphate-dependent amino acid racemases of broad substrate specificity catalyze transamination as a side-reaction. We studied the stereospecificities for hydrogen abstraction from C-4’ of the bound pyridoxamine 5’-phosphate during transamination from pyridoxamine 5’-phosphate to pyruvate catalyzed by three amino acid racemases of broad substrate specificity. When the enzymes were incubated with (4’S)- or (4’R)-[4’-3H]- pyridoxamine 5’-phosphate in the presence of pyruvate, tritium was released into the solvent from both pyridoxamine 5’-phosphates. Thus, these enzymes abstract a hydrogen non-stereospecifically from C-4’ of the coenzyme in contrast to the other pyridoxal 5’-phosphate-dependent enzymes so far studied which catalyze the stereospecific hydrogen removal. Amino acid racemase of broad substrate specificity from Pseudomonas putida produced D- and L-glutamate from α-ketoglutarate through the transamination with L-ornithine. Because glutamate does not serve as a substrate for racemization, the enzyme catalyzed the non-stereospecific overall transamination between L-ornithine and α-ketoglutarate. -

Letters to Nature
letters to nature Received 7 July; accepted 21 September 1998. 26. Tronrud, D. E. Conjugate-direction minimization: an improved method for the re®nement of macromolecules. Acta Crystallogr. A 48, 912±916 (1992). 1. Dalbey, R. E., Lively, M. O., Bron, S. & van Dijl, J. M. The chemistry and enzymology of the type 1 27. Wolfe, P. B., Wickner, W. & Goodman, J. M. Sequence of the leader peptidase gene of Escherichia coli signal peptidases. Protein Sci. 6, 1129±1138 (1997). and the orientation of leader peptidase in the bacterial envelope. J. Biol. Chem. 258, 12073±12080 2. Kuo, D. W. et al. Escherichia coli leader peptidase: production of an active form lacking a requirement (1983). for detergent and development of peptide substrates. Arch. Biochem. Biophys. 303, 274±280 (1993). 28. Kraulis, P.G. Molscript: a program to produce both detailed and schematic plots of protein structures. 3. Tschantz, W. R. et al. Characterization of a soluble, catalytically active form of Escherichia coli leader J. Appl. Crystallogr. 24, 946±950 (1991). peptidase: requirement of detergent or phospholipid for optimal activity. Biochemistry 34, 3935±3941 29. Nicholls, A., Sharp, K. A. & Honig, B. Protein folding and association: insights from the interfacial and (1995). the thermodynamic properties of hydrocarbons. Proteins Struct. Funct. Genet. 11, 281±296 (1991). 4. Allsop, A. E. et al.inAnti-Infectives, Recent Advances in Chemistry and Structure-Activity Relationships 30. Meritt, E. A. & Bacon, D. J. Raster3D: photorealistic molecular graphics. Methods Enzymol. 277, 505± (eds Bently, P. H. & O'Hanlon, P. J.) 61±72 (R. Soc. Chem., Cambridge, 1997). -

Generated by SRI International Pathway Tools Version 25.0, Authors S
Authors: Pallavi Subhraveti Ron Caspi Quang Ong Peter D Karp An online version of this diagram is available at BioCyc.org. Biosynthetic pathways are positioned in the left of the cytoplasm, degradative pathways on the right, and reactions not assigned to any pathway are in the far right of the cytoplasm. Transporters and membrane proteins are shown on the membrane. Ingrid Keseler Periplasmic (where appropriate) and extracellular reactions and proteins may also be shown. Pathways are colored according to their cellular function. Gcf_000725805Cyc: Streptomyces xanthophaeus Cellular Overview Connections between pathways are omitted for legibility. -

Exploring the Chemistry and Evolution of the Isomerases
Exploring the chemistry and evolution of the isomerases Sergio Martínez Cuestaa, Syed Asad Rahmana, and Janet M. Thorntona,1 aEuropean Molecular Biology Laboratory, European Bioinformatics Institute, Wellcome Trust Genome Campus, Hinxton, Cambridge CB10 1SD, United Kingdom Edited by Gregory A. Petsko, Weill Cornell Medical College, New York, NY, and approved January 12, 2016 (received for review May 14, 2015) Isomerization reactions are fundamental in biology, and isomers identifier serves as a bridge between biochemical data and ge- usually differ in their biological role and pharmacological effects. nomic sequences allowing the assignment of enzymatic activity to In this study, we have cataloged the isomerization reactions known genes and proteins in the functional annotation of genomes. to occur in biology using a combination of manual and computa- Isomerases represent one of the six EC classes and are subdivided tional approaches. This method provides a robust basis for compar- into six subclasses, 17 sub-subclasses, and 245 EC numbers cor- A ison and clustering of the reactions into classes. Comparing our responding to around 300 biochemical reactions (Fig. 1 ). results with the Enzyme Commission (EC) classification, the standard Although the catalytic mechanisms of isomerases have already approach to represent enzyme function on the basis of the overall been partially investigated (3, 12, 13), with the flood of new data, an integrated overview of the chemistry of isomerization in bi- chemistry of the catalyzed reaction, expands our understanding of ology is timely. This study combines manual examination of the the biochemistry of isomerization. The grouping of reactions in- chemistry and structures of isomerases with recent developments volving stereoisomerism is straightforward with two distinct types cis-trans in the automatic search and comparison of reactions. -

Taxonomic and Functional Analyses of the Supragingival Microbiome from Caries-Affected and Caries-Free Hosts
Taxonomic and Functional Analyses of the Supragingival Microbiome from Caries-Affected and Caries-Free Hosts Jinzhi He1, Qichao Tu2,3, Yichen Ge1, Yujia Qin3, Bomiao Cui1, Xiaoyu Hu1, Yuxia Wang1, Ye Deng4, Kun Wang1, Joy D. Van Nostrand3, Jiyao Li 1, Jizhong Zhou3,5,6, Yan Li 1, Xuedong Zhou1 1 State Key Laboratory of Oral Diseases, National Clinical Research Center for Oral Diseases, West China Hospital of Stomatology, Sichuan University, Chengdu, China 2 Department of Marine Sciences, Ocean College, Zhejiang University, Hangzhou, China 3 Institute for Environmental Genomics, Department of Microbiology and Plant Biology, and School of Civil Engineering and Environmental Sciences, University of Oklahoma, Norman, USA 4 Research Center for Eco-Environmental Sciences, Chinese Academy of Sciences, Beijing, China 5 State Key Joint Laboratory of Environment Simulation and Pollution Control, School of Environment, Tsinghua University, Beijing, China 6 Earth Science Division, Lawrence Berkeley National Laboratory, Berkeley, USA Abstract Caries is one of the most prevalent and costly infectious diseases affecting humans of all ages. It is initiated by cariogenic supragingival dental plaques forming on salivacoated tooth surfaces, yet the etiology remains elusive. To determine which microbial populations may predispose a patient to caries, we report here an in-depth and comprehensive view of the microbial community associated with supragingival dental plaque collected from the healthy teeth of caries patients and healthy adults. We found that microbial communities from caries patients had a higher evenness and inter-individual variations but simpler ecological networks compared to healthy controls despite the overall taxonomic structure being similar. Genera including Selenomonas, Treponema, Atopobium, and Bergeriella were distributed differently between the caries and healthy groups with disturbed co- occurrence patterns. -

K. ENZYME KINETICS and CATALYSIS Basic Kinetics
K. ENZYME KINETICS AND CATALYSIS Biochemical reactions are too slow to support life at room temperature. Whether it is the synthesis of proteins, the metabolism of sugars or the oxidation of fatty acids, all of it proceeds on a time scale unsuitable for life. Moreover, many reactions that are undesirable proceed on a comparable timescale, making it even more difficult to coordinate the necessary chemistry to support life. How useful would it be to have polynucleotides degrading faster than they are synthesized? The basic reactions associated with the synthesis and degradation of essential polymers, and with the metabolism of nutrients all require massive rate accelerations to be of any service to the cell. By specifically accelerating those reactions of interest without accidentally accelerating undesired reactions, as would happen by simply heating the pot, biochemical catalysts shape chemistry in such a way as to permit every biological process required for survival and propagation. Basic Kinetics Reaction Rates The rate of a reaction is defined as the instantaneous rate of change in the concentration of a starting material (“substrate” in biochemical nomenclature – abbreviated “S”) or a product with respect to time. By definition, reaction rates are always positive, which leads to the following definition of reaction rate, abbreviated v (for velocity) in most of the biochemical literature. S → P (eq. K.1) d[P] d[S] v = = − (eq. K.2) dt dt Because these are instantaneous rates of change, they can be obtained as tangents to curves that plot [P] or [S] vs. time. Typically, one takes the initial rate (Figure K.1) – as close to the point of € initiation of the reaction as possible, since the reaction components are well-defined at that moment. -

Is D-Aspartate Produced by Glutamic-Oxaloacetic Transaminase-1 Like 1 (Got1l1): a Putative Aspartate Racemase?
Amino Acids (2015) 47:79–86 DOI 10.1007/s00726-014-1847-3 ORIGINAL ARTICLE Is d-aspartate produced by glutamic-oxaloacetic transaminase-1 like 1 (Got1l1): a putative aspartate racemase? Ayumi Tanaka-Hayashi · Shuuhei Hayashi · Ran Inoue · Tomokazu Ito · Kohtarou Konno · Tomoyuki Yoshida · Masahiko Watanabe · Tohru Yoshimura · Hisashi Mori Received: 23 July 2014 / Accepted: 25 September 2014 / Published online: 7 October 2014 © The Author(s) 2014. This article is published with open access at Springerlink.com Abstract D-Aspartate is an endogenous free amino acid hippocampus. The recombinant Got1l1 expressed in mam- in the brain, endocrine tissues, and exocrine tissues in malian cells showed L-aspartate aminotransferase activity, mammals, and it plays several physiological roles. In the but lacked aspartate racemase activity. These findings sug- testis, D-aspartate is detected in elongate spermatids, Ley- gest that Got1l1 is not the major aspartate racemase and dig cells, and Sertoli cells, and implicated in the synthesis there might be an as yet unknown D-aspartate-synthesizing and release of testosterone. In the hippocampus, D-aspartate enzyme. strongly enhances N-methyl-D-aspartate receptor-depend- ent long-term potentiation and is involved in learning and Keywords Glutamic-oxaloacetic transaminase-1 like 1 · memory. The existence of aspartate racemase, a candidate D-Aspartate · Knockout mice · Testis · Hippocampus · enzyme for D-aspartate production, has been suggested. Recombinant protein expression Recently, mouse glutamic-oxaloacetic transaminase 1-like 1 (Got1l1) has been reported to synthesize substantially Abbreviations D-aspartate from L-aspartate and to be involved in adult Got1l1 Glutamic-oxaloacetic transaminase-1 like 1 neurogenesis. -

Bacterial Selenoproteins: a Role in Pathogenesis and Targets for Antimicrobial Development
University of Central Florida STARS Electronic Theses and Dissertations, 2004-2019 2009 Bacterial Selenoproteins: A Role In Pathogenesis And Targets For Antimicrobial Development Sarah Rosario University of Central Florida Part of the Medical Sciences Commons Find similar works at: https://stars.library.ucf.edu/etd University of Central Florida Libraries http://library.ucf.edu This Doctoral Dissertation (Open Access) is brought to you for free and open access by STARS. It has been accepted for inclusion in Electronic Theses and Dissertations, 2004-2019 by an authorized administrator of STARS. For more information, please contact [email protected]. STARS Citation Rosario, Sarah, "Bacterial Selenoproteins: A Role In Pathogenesis And Targets For Antimicrobial Development" (2009). Electronic Theses and Dissertations, 2004-2019. 3822. https://stars.library.ucf.edu/etd/3822 BACTERIAL SELENOPROTEINS: A ROLE IN PATHOGENESIS AND TARGETS FOR ANTIMICROBIAL DEVELOPMENT. by SARAH E. ROSARIO B.S. Florida State University, 2000 M.P.H. University of South Florida, 2002 A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Burnett School of Biomedical Sciences in the College of Medicine at the University of Central Florida Orlando, Florida Summer Term 2009 Major Professor: William T. Self © 2009 Sarah E. Rosario ii ABSTRACT Selenoproteins are unique proteins in which selenocysteine is inserted into the polypeptide chain by highly specialized translational machinery. They exist within all three kingdoms of life. The functions of these proteins in biology are still being defined. In particular, the importance of selenoproteins in pathogenic microorganisms has received little attention. We first established that a nosocomial pathogen, Clostridium difficile, utilizes a selenoenzyme dependent pathway for energy metabolism. -

Designed Mono- and Di-Covalent Inhibitors Trap Modeled Functional Motions for Trypanosoma Cruzi Proline Racemase in Crystallography
RESEARCH ARTICLE Designed mono- and di-covalent inhibitors trap modeled functional motions for Trypanosoma cruzi proline racemase in crystallography 1¤a 2¤b 2 Patricia de Aguiar Amaral , Delphine Autheman , Guilherme Dias de MeloID , 1 1 2¤c 2¤d Nicolas Gouault , Jean-FrancËois Cupif , Sophie GoyardID , Patricia Dutra , 2 2 3 4 4 Nicolas Coatnoan , Alain Cosson , Damien Monet , Frederick Saul , Ahmed HaouzID , 1 3 2¤e a1111111111 Philippe Uriac *, Arnaud Blondel *, Paola MinoprioID * a1111111111 a1111111111 1 Universite de Rennes 1, Equipe Chimie organique et interfaces (CORINT), UMR 6226 Sciences Chimiques de Rennes, Rennes, France, 2 Institut Pasteur, Laboratoire des Processus Infectieux à TrypanosomatideÂs, a1111111111 DeÂpartement Infection et EpideÂmiologie, Paris, France, 3 Institut Pasteur, Unite de Bioinformatique a1111111111 Structurale, DeÂpartement de Biologie Structurale et Chimie, CNRS-UMR 3528, Paris, France, 4 Institut Pasteur, Plateforme de Cristallographie, DeÂpartement de Biologie Structurale et Chimie, CNRS-UMR 3528, Paris, France ¤a Current address: LaPlaM/PPGCA, Universidade do Extremo Sul Catarinense, CriciuÂmaÐSC, Brazil ¤b Current address: Cell Surface Signalling Laboratory, Wellcome Trust Sanger Institute, Wellcome Trust OPEN ACCESS Genome Campus, Cambridge, United Kingdom Citation: Amaral PdA, Autheman D, de Melo GD, ¤c Current address: Institut Pasteur, Centre d'Innovation et Recherche Technologique, Paris, France Gouault N, Cupif J-F, Goyard S, et al. (2018) ¤d Current address: LaboratoÂrio de BioquõÂmica de ProtozoaÂrios e Imunofisiologia do ExercõÂcio, Departamento de Microbiologia, Imunologia e Parasitologia, Universidade do Estado do Rio de Janeiro, Rio Designed mono- and di-covalent inhibitors trap de Janeiro, Brazil modeled functional motions for Trypanosoma cruzi ¤e Current address: Trypanosomatids Infectious Processes Laboratory, Scientific Platform Pasteur-USP, proline racemase in crystallography.