Taxonomic and Functional Analyses of the Supragingival Microbiome from Caries-Affected and Caries-Free Hosts
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Fulltext01.Pdf
http://www.diva-portal.org This is the published version of a paper published in Cellular and Molecular Life Sciences (CMLS). Citation for the original published paper (version of record): Cava, F., Lam, H., de Pedro, M., Waldor, M. (2011) Emerging knowledge of regulatory roles of D-amino acids in bacteria. Cellular and Molecular Life Sciences (CMLS), 68(5): 817-831 http://dx.doi.org/10.1007/s00018-010-0571-8 Access to the published version may require subscription. N.B. When citing this work, cite the original published paper. Permanent link to this version: http://urn.kb.se/resolve?urn=urn:nbn:se:umu:diva-81861 Cell. Mol. Life Sci. (2011) 68:817–831 DOI 10.1007/s00018-010-0571-8 Cellular and Molecular Life Sciences REVIEW Emerging knowledge of regulatory roles of D-amino acids in bacteria Felipe Cava • Hubert Lam • Miguel A. de Pedro • Matthew K. Waldor Received: 13 July 2010 / Revised: 24 September 2010 / Accepted: 14 October 2010 / Published online: 14 December 2010 Ó The Author(s) 2010. This article is published with open access at Springerlink.com Abstract The D-enantiomers of amino acids have been Keywords D-amino acid Á Racemase Á Stationary phase Á thought to have relatively minor functions in biological Peptidoglycan Á Biofilm Á Regulation processes. While L-amino acids clearly predominate in nat- ure, D-amino acids are sometimes found in proteins that are Abbreviations not synthesized by ribosomes, and D-Ala and D-Glu are NRP Nonribosomal peptide routinely found in the peptidoglycan cell wall of bacteria. PG Peptidoglycan Here, we review recent findings showing that D-amino acids GlcNAc N-acetyl glucosamine have previously unappreciated regulatory roles in the bac- MurNAc N-acetylmuramic acid terial kingdom. -

Title Non-Stereospecific Transamination Catalyzed by Pyridoxal Phosphate-Dependent Amino Acid Racemases of Broad Substrate Speci
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Kyoto University Research Information Repository Non-stereospecific Transamination Catalyzed by Pyridoxal Phosphate-dependent Amino Acid Racemases of Broad Title Substrate Specificity (MOLECULAR BIOFUNCTION- Molecular Microbial Science) Esaki, Nobuyoshi; Yoshimura, Tohru; Soda, Kenji; Lim, Author(s) Young Hee Citation ICR annual report (1999), 5: 46-47 Issue Date 1999-03 URL http://hdl.handle.net/2433/65185 Right Type Article Textversion publisher Kyoto University 46 ICR Annual Report, Vol. 5, 1998 Non-stereospecific Transamination Catalyzed by Pyridoxal Phosphate-dependent Amino Acid Racemases of Broad Substrate Specificity Nobuyoshi Esaki, Tohru Yoshimura, Kenji Soda and Young Hee Lim Pyridoxal 5’-phosphate-dependent amino acid racemases of broad substrate specificity catalyze transamination as a side-reaction. We studied the stereospecificities for hydrogen abstraction from C-4’ of the bound pyridoxamine 5’-phosphate during transamination from pyridoxamine 5’-phosphate to pyruvate catalyzed by three amino acid racemases of broad substrate specificity. When the enzymes were incubated with (4’S)- or (4’R)-[4’-3H]- pyridoxamine 5’-phosphate in the presence of pyruvate, tritium was released into the solvent from both pyridoxamine 5’-phosphates. Thus, these enzymes abstract a hydrogen non-stereospecifically from C-4’ of the coenzyme in contrast to the other pyridoxal 5’-phosphate-dependent enzymes so far studied which catalyze the stereospecific hydrogen removal. Amino acid racemase of broad substrate specificity from Pseudomonas putida produced D- and L-glutamate from α-ketoglutarate through the transamination with L-ornithine. Because glutamate does not serve as a substrate for racemization, the enzyme catalyzed the non-stereospecific overall transamination between L-ornithine and α-ketoglutarate. -

Is D-Aspartate Produced by Glutamic-Oxaloacetic Transaminase-1 Like 1 (Got1l1): a Putative Aspartate Racemase?
Amino Acids (2015) 47:79–86 DOI 10.1007/s00726-014-1847-3 ORIGINAL ARTICLE Is d-aspartate produced by glutamic-oxaloacetic transaminase-1 like 1 (Got1l1): a putative aspartate racemase? Ayumi Tanaka-Hayashi · Shuuhei Hayashi · Ran Inoue · Tomokazu Ito · Kohtarou Konno · Tomoyuki Yoshida · Masahiko Watanabe · Tohru Yoshimura · Hisashi Mori Received: 23 July 2014 / Accepted: 25 September 2014 / Published online: 7 October 2014 © The Author(s) 2014. This article is published with open access at Springerlink.com Abstract D-Aspartate is an endogenous free amino acid hippocampus. The recombinant Got1l1 expressed in mam- in the brain, endocrine tissues, and exocrine tissues in malian cells showed L-aspartate aminotransferase activity, mammals, and it plays several physiological roles. In the but lacked aspartate racemase activity. These findings sug- testis, D-aspartate is detected in elongate spermatids, Ley- gest that Got1l1 is not the major aspartate racemase and dig cells, and Sertoli cells, and implicated in the synthesis there might be an as yet unknown D-aspartate-synthesizing and release of testosterone. In the hippocampus, D-aspartate enzyme. strongly enhances N-methyl-D-aspartate receptor-depend- ent long-term potentiation and is involved in learning and Keywords Glutamic-oxaloacetic transaminase-1 like 1 · memory. The existence of aspartate racemase, a candidate D-Aspartate · Knockout mice · Testis · Hippocampus · enzyme for D-aspartate production, has been suggested. Recombinant protein expression Recently, mouse glutamic-oxaloacetic transaminase 1-like 1 (Got1l1) has been reported to synthesize substantially Abbreviations D-aspartate from L-aspartate and to be involved in adult Got1l1 Glutamic-oxaloacetic transaminase-1 like 1 neurogenesis. -

SI Appendix Index 1
SI Appendix Index Calculating chemical attributes using EC-BLAST ................................................................................ 2 Chemical attributes in isomerase reactions ............................................................................................ 3 Bond changes …..................................................................................................................................... 3 Reaction centres …................................................................................................................................. 5 Substrates and products …..................................................................................................................... 6 Comparative analysis …........................................................................................................................ 7 Racemases and epimerases (EC 5.1) ….................................................................................................. 7 Intramolecular oxidoreductases (EC 5.3) …........................................................................................... 8 Intramolecular transferases (EC 5.4) ….................................................................................................. 9 Supporting references …....................................................................................................................... 10 Fig. S1. Overview …............................................................................................................................ -

Aspartate Racemase, Generating Neuronal D-Aspartate, Regulates Adult Neurogenesis
Aspartate racemase, generating neuronal D-aspartate, regulates adult neurogenesis Paul M. Kima, Xin Duana,b,1, Alex S. Huanga, Cindy Y. Liub, Guo-li Minga,b,c, Hongjun Songa,b,c,2 and Solomon H. Snydera,d,e,2 aThe Solomon H. Snyder Department of Neuroscience, bInstitute for Cell Engineering, cDepartment of Neurology, dDepartment of Pharmacology and Molecular Sciences and eDepartment of Psychiatry and Behavioral Sciences, The Johns Hopkins University School of Medicine, Baltimore, MD 21205 Contributed by Solomon Snyder, December 24, 2009 (sent for review December 3, 2009) D-Aspartic acid is abundant in the developing brain. We have identified interfering with substrate induced closure of the enzyme’s active site and cloned mammalian aspartate racemase (DR), which converts L- and enhances racemization (18). aspartate to D-aspartate and colocalizes with D-aspartate in the brain In a NCBI protein database search, we discovered a putative and neuroendocrine tissues. Depletion of DR by retrovirus-mediated DR, a GOT-like protein in which GOT’s tryptophan-140 is re- expression of short-hairpin RNA in newborn neurons of the adult placed by lysine and arginine-292 is substituted with a glutamine, hippocampus elicits profound defects in the dendritic development suggesting possibe racemase activity (Fig. S1). Similar to the pre- and survival of newborn neurons and survival. Because D-aspartate is a viously mentioned tryptophan-140–to-histidine mutational anal- potential endogenous ligand for NMDA receptors, the loss of which ysis of GOT, lysine has a proton that can be directly donated for elicits a phenotype resembling DR depletion, D-aspartate may function racemization (Fig. -

Α-Methylacyl-Coa Racemase and Argininosuccinate Lyase
A460etukansi.fm Page 1 Friday, May 26, 2006 11:40 AM A 460 OULU 2006 A 460 UNIVERSITY OF OULU P.O. Box 7500 FI-90014 UNIVERSITY OF OULU FINLAND ACTA UNIVERSITATIS OULUENSIS ACTA UNIVERSITATIS OULUENSIS ACTA A SERIES EDITORS SCIENTIAE RERUM Prasenjit Bhaumik NATURALIUM Prasenjit Bhaumik Prasenjit ASCIENTIAE RERUM NATURALIUM Professor Mikko Siponen PROTEIN CRYSTALLOGRAPHIC BHUMANIORA STUDIES TO UNDERSTAND Professor Harri Mantila THE REACTION MECHANISM CTECHNICA Professor Juha Kostamovaara OF ENZYMES: DMEDICA α Professor Olli Vuolteenaho -METHYLACYL-CoA RACEMASE AND ESCIENTIAE RERUM SOCIALIUM Senior assistant Timo Latomaa ARGININOSUCCINATE LYASE FSCRIPTA ACADEMICA Communications Officer Elna Stjerna GOECONOMICA Senior Lecturer Seppo Eriksson EDITOR IN CHIEF Professor Olli Vuolteenaho EDITORIAL SECRETARY Publication Editor Kirsti Nurkkala FACULTY OF SCIENCE, DEPARTMENT OF BIOCHEMISTRY, BIOCENTER OULU, ISBN 951-42-8090-3 (Paperback) UNIVERSITY OF OULU ISBN 951-42-8091-1 (PDF) ISSN 0355-3191 (Print) ISSN 1796-220X (Online) ACTA UNIVERSITATIS OULUENSIS A Scientiae Rerum Naturalium 460 PRASENJIT BHAUMIK PROTEIN CRYSTALLOGRAPHIC STUDIES TO UNDERSTAND THE REACTION MECHANISM OF ENZYMES: α-METHYLACYL-COA RACEMASE AND ARGININOSUCCINATE LYASE Academic Dissertation to be presented with the assent of the Faculty of Science, University of Oulu, for public discussion in Kuusamonsali (Auditorium YB210), Linnanmaa, on June 6th, 2006, at 12 noon OULUN YLIOPISTO, OULU 2006 Copyright © 2006 Acta Univ. Oul. A 460, 2006 Supervised by Professor Rik Wierenga Reviewed -

D-Amino Acids in Health and Disease: a Focus on Cancer
nutrients Review d-amino Acids in Health and Disease: A Focus on Cancer Jacco J.A.J. Bastings 1,2, Hans M. van Eijk 1, Steven W. Olde Damink 1,3 and Sander S. Rensen 1,* 1 Department of Surgery, NUTRIM School of Nutrition and Translational Research in Metabolism, Maastricht University, 6200 MD Maastricht, The Netherlands; [email protected] (J.J.A.J.B.); [email protected] (H.M.v.E.); [email protected] (S.W.O.D.) 2 Department of Human Biology, NUTRIM School of Nutrition and Translational Research in Metabolism, Maastricht University, 6200 MD Maastricht, The Netherlands 3 Department of General, Visceral and Transplantation Surgery, RWTH University Hospital Aachen, 52074 Aachen, Germany * Correspondence: [email protected] Received: 29 July 2019; Accepted: 9 September 2019; Published: 12 September 2019 Abstract: d-amino acids, the enantiomeric counterparts of l-amino acids, were long considered to be non-functional or not even present in living organisms. Nowadays, d-amino acids are acknowledged to play important roles in numerous physiological processes in the human body. The most commonly studied link between d-amino acids and human physiology concerns the contribution of d-serine and d-aspartate to neurotransmission. These d-amino acids and several others have also been implicated in regulating innate immunity and gut barrier function. Importantly, the presence of certain d-amino acids in the human body has been linked to several diseases including schizophrenia, amyotrophic lateral sclerosis, and age-related disorders such as cataract and atherosclerosis. Furthermore, increasing evidence supports a role for d-amino acids in the development, pathophysiology, and treatment of cancer. -
![Viewed in (104)]](https://docslib.b-cdn.net/cover/1427/viewed-in-104-3121427.webp)
Viewed in (104)]
The Role in Translation of Editing and Multi-Synthetase Complex Formation by Aminoacyl-tRNA Synthetases Dissertation Presented in Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy in the Graduate School of The Ohio State University By Medha Vijay Raina, M.Sc. Ohio State Biochemistry Graduate Program The Ohio State University 2014 Dissertation Committee: Dr. Michael Ibba, Advisor Dr. Juan Alfonzo Dr. Irina Artsimovitch Dr. Kurt Fredrick Dr. Karin Musier-Forsyth Copyright by Medha Vijay Raina 2014 ABSTRACT Aminoacyl-tRNA synthetases (aaRSs) catalyze the first step of translation, aminoacylation. These enzymes attach amino acids (aa) to their cognate tRNAs to form aminoacyl-tRNA (aa-tRNA), an important substrate in protein synthesis, which is delivered to the ribosome as a ternary complex with translation elongation factor 1A (EF1A) and GTP. All aaRSs have an aminoacylation domain, which is the active site that recognizes the specific amino acid, ATP, and the 3′ end of the bound tRNA to catalyze the aminoacylation reaction. Apart from the aminoacylation domain, some aaRSs have evolved additional domains that are involved in interacting with other proteins, recognizing and binding the tRNA anticodon, and editing misacylated tRNA thereby expanding their role in and beyond translation. One such function of the aaRS is to form a variety of complexes with each other and with other factors by interacting via additional N or C terminal extensions. For example, several archaeal and eukaryotic aaRSs are known to associate with EF1A or other aaRSs forming higher order complexes, although the role of these multi-synthetase complexes (MSC) in translation remains largely unknown. -
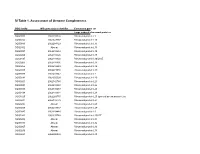
SI Table 1. Assessment of Genome Completeness
SI Table 1. Assessment of Genome Completeness COG family IMG gene object identifier Conserved gene set Large subunit ribosomal proteins COG0081 2062288324 Ribosomal protein L1 COG0244 2062347387 Ribosomal protein L10 COG0080 2062288323 Ribosomal protein L11 COG0102 Absent Ribosomal protein L13 COG0093 2062418832 Ribosomal protein L14 COG0200 2062418826 Ribosomal protein L15 COG0197 2062418838 Ribosomal protein L16/L10E COG0203 2062418836 Ribosomal protein L17 COG0256 2062418829 Ribosomal protein L18 COG0335 2062273558 Ribosomal protein L19 COG0090 2062418842 Ribosomal protein L2 COG0292 2062350539 Ribosomal protein L20 COG0261 2062142780 Ribosomal protein L21 COG0091 2062418840 Ribosomal protein L22 COG0089 2062138283 Ribosomal protein L23 COG0198 2062418834 Ribosomal protein L24 COG1825 2062269715 Ribosomal protein L25 (general stress protein Ctc) COG0211 2062142779 Ribosomal protein L27 COG0227 Absent Ribosomal protein L28 COG0255 2062418837 Ribosomal protein L29 COG0087 2062154483 Ribosomal protein L3 COG1841 2062335748 Ribosomal protein L30/L7E COG0254 Absent Ribosomal protein L31 COG0333 Absent Ribosomal protein L32 COG0267 Absent Ribosomal protein L33 COG0230 Absent Ribosomal protein L34 COG0291 2062350538 Ribosomal protein L35 COG0257 Absent Ribosomal protein L36 COG0088 2062138282 Ribosomal protein L4 COG0094 2062418833 Ribosomal protein L5 COG0097 2062418830 Ribosomal protein L6P/L9E COG0222 2062288326 Ribosomal protein L7/L12 COG0359 2062209880 Ribosomal protein L9 Small subunit ribosomal proteins COG0539 Absent Ribosomal protein -

Drug Discovery Targeting Amino Acid Racemases Paola Conti, Lucia Tamborini, Andrea Pinto, Arnaud Blondel, Paola Minoprio, Andrea Mozzarelli, Carlo De Micheli
Drug Discovery Targeting Amino Acid Racemases Paola Conti, Lucia Tamborini, Andrea Pinto, Arnaud Blondel, Paola Minoprio, Andrea Mozzarelli, Carlo de Micheli To cite this version: Paola Conti, Lucia Tamborini, Andrea Pinto, Arnaud Blondel, Paola Minoprio, et al.. Drug Discovery Targeting Amino Acid Racemases. Chemical Reviews, American Chemical Society, 2011, 111 (11), pp.6919-6946. 10.1021/cr2000702. pasteur-02510858 HAL Id: pasteur-02510858 https://hal-pasteur.archives-ouvertes.fr/pasteur-02510858 Submitted on 7 Apr 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Drug Discovery Targeting Amino Acid Racemases Paola Conti,a Lucia Tamborini, a Andrea Pinto,a Arnaud Blondel,b Paola Minoprio,c Andrea Mozzarellid and Carlo De Micheli a* aDipartimento di Scienze Farmaceutiche ‘P. Pratesi’, via Mangiagalli 25, 20133 Milano, Italy; bUnité de Bioinformatique Structurale, CNRS-URA 2185, Département de Biologie Structurale et Chimie, 25 rue du Dr. Roux, 75724 Paris, France. cInstitut Pasteur, Laboratoire des Processus Infectieux à Trypanosoma; Départment d’Infection and Epidémiologie; 25 rue du Dr. Roux, 75724 Paris, France dDipartimento di Biochimica e Biologia Molecolare, via G. P. Usberti 23/A, 43100 Parma, Italy and Istituto di Biostrutture e Biosistemi, viale Medaglie d’oro, Rome, Italy. -

D-Aspartate Consumption Selectively Promotes Intermediate-Term Spatial
www.nature.com/scientificreports OPEN d‑Aspartate consumption selectively promotes intermediate‑term spatial memory and the expression of hippocampal NMDA receptor subunits Gergely Zachar 1,2*, Róbert Kemecsei1,2, Szilvia Márta Papp1, Katalin Wéber1, Tamás Kisparti1, Teadora Tyler1, Gábor Gáspár1, Tamás Balázsa1 & András Csillag1 d‑Aspartate (d‑Asp) and d‑serine (d‑Ser) have been proposed to promote early‑phase LTP in vitro and to enhance spatial memory in vivo. Here, we investigated the behavioural efects of chronic consumption of d‑Asp and d‑Ser on spatial learning of mice together with the expression of NMDA receptors. We also studied the alterations of neurogenesis by morphometric analysis of bromo‑ deoxyuridine incorporating and doublecortin expressing cells in the hippocampus. Our results specify a time period (3–4 h post‑training), within which the animals exposed to d‑Asp (but not d‑Ser) show a more stable memory during retrieval. The cognitive improvement is due to elimination of transient bouts of destabilization and reconsolidation of memory, rather than to enhanced acquisition. d‑Asp also protracted reversal learning probably due to reduced plasticity. Expression of GluN1 and GluN2A subunits was elevated in the hippocampus of d‑Asp (but not d‑Ser) treated mice. d‑Asp or d‑Ser did not alter the proliferation of neuronal progenitor cells in the hippocampus. The observed learning‑ related changes evoked by d‑Asp are unlikely to be due to enhanced proliferation and recruitment of new neurones. Rather, they are likely associated with an upregulation of NMDA receptors, as well as a reorganization of receptor subunit assemblies in existing hippocampal/dentate neurons. -

The Role of Amino Acids in Neurotransmission and Fluorescent Tools for Their Detection
International Journal of Molecular Sciences Review The Role of Amino Acids in Neurotransmission and Fluorescent Tools for Their Detection Rochelin Dalangin 1, Anna Kim 1 and Robert E. Campbell 1,2,* 1 Department of Chemistry, University of Alberta, Edmonton, AB T6G 2G2, Canada; [email protected] (R.D.); [email protected] (A.K.) 2 Department of Chemistry, Graduate School of Science, The University of Tokyo, Bunkyo City, Tokyo 113-0033, Japan * Correspondence: [email protected]; Tel.: +1-7804921849 Received: 29 July 2020; Accepted: 24 August 2020; Published: 27 August 2020 Abstract: Neurotransmission between neurons, which can occur over the span of a few milliseconds, relies on the controlled release of small molecule neurotransmitters, many of which are amino acids. Fluorescence imaging provides the necessary speed to follow these events and has emerged as a powerful technique for investigating neurotransmission. In this review, we highlight some of the roles of the 20 canonical amino acids, GABA and β-alanine in neurotransmission. We also discuss available fluorescence-based probes for amino acids that have been shown to be compatible for live cell imaging, namely those based on synthetic dyes, nanostructures (quantum dots and nanotubes), and genetically encoded components. We aim to provide tool developers with information that may guide future engineering efforts and tool users with information regarding existing indicators to facilitate studies of amino acid dynamics. Keywords: amino acids; neurotransmission; fluorescence; imaging; biosensors; neurotransmitters; indicators 1. Introduction Neurons communicate to each other by the release of chemicals stored in synaptic vesicles across specialized gaps known as synapses. These chemicals diffuse across the synapse and bind to their target receptors on adjacent neurons to modulate their physiological states.