NIH Public Access Author Manuscript Curr Opin Chem Biol
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Arginine Enzymatic Deprivation and Diet Restriction for Cancer Treatment
Brazilian Journal of Pharmaceutical Sciences Review http://dx.doi.org/10.1590/s2175-97902017000300200 Arginine enzymatic deprivation and diet restriction for cancer treatment Wissam Zam* Al-Andalus University for Medical Sciences, Faculty of Pharmacy, Analytical and Food Chemistry, Tartous, Syrian Arab Republic Recent findings in amino acid metabolism and the differences between normal, healthy cells and neoplastic cells have revealed that targeting single amino acid metabolic enzymes in cancer therapy is a promising strategy for the development of novel therapeutic agents. Arginine is derived from dietary protein intake, body protein breakdown, or endogenous de novo arginine production and several studies have revealed disturbances in its synthesis and metabolism which could enhance or inhibit tumor cell growth. Consequently, there has been an increased interest in the arginine-depleting enzymes and dietary deprivation of arginine and its precursors as a potential antineoplastic therapy. This review outlines the most recent advances in targeting arginine metabolic pathways in cancer therapy and the different chemo- and radio-therapeutic approaches to be co-applied. Key words: Arginine-depleting enzyme/antineoplastic therapy. Dietary deprivation. INTRODUCTION variety of human cancer cells have been found to be auxotrophic for arginine, depletion of which results in Certain cancers may be auxotrophic for a particular cell death (Tytell, Neuman, 1960; Kraemer, 1964; Dillon amino acid, and amino acid deprivation is one method to et al., 2004). Arginine can be degraded by three enzymes: treat these tumors. The strategy of enzymatic degradation arginase, arginine decarboxylase and arginine deiminase of amino acids to deprive malignant cells of important (ADI). Both arginine decarboxylase and ADI are not nutrients is an established component of induction therapy expressed in mammalian cells (Morris, 2007; Miyazaki of several tumor cells. -
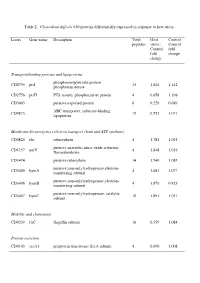
Table 2: Clostridium Difficile 630 Proteins Differentially Expressed in Response to Heat Stress
Table 2: Clostridium difficile 630 proteins differentially expressed in response to heat stress. Locus Gene name Description Total Heat Control : peptides stress : Control Control fold fold change change Transport/binding proteins and lipoproteins phosphoenolpyruvate-protein CD2755 ptsI 15 1.644 1.142 phosphotransferase CD2756 ptsH PTS system, phosphocarrier protein 4 0.658 1.198 CD3043 putative exported protein 6 0.229 0.689 ABC transporter, substrate-binding CD0873 15 0.521 1.071 lipoprotein Membrane bioenergetics (electron transport chain and ATP synthase) CD0825 rbr rubrerythrin 4 1.781 1.035 putative anaerobic nitric oxide reductase CD1157 norV 4 1.848 1.016 flavorubredoxin CD1474 putative ruberythrin 14 1.540 1.085 putative iron-only hydrogenase,electron- CD3405 hymA 4 1.681 1.077 transferring subunit putative iron-only hydrogenase,electron- CD3406 hymB 4 1.870 0.923 transferring subunit putative iron-only hydrogenase, catalytic CD3407 hymC 10 1.891 1.051 subunit Mobility and chemotaxis CD0239 fliC flagellin subunit 16 0.559 1.084 Protein secretion CD0143 secA1 preprotein translocase SecA subunit 4 0.690 1.008 Specific pathways putative oxidoreductase, thiamine diP-binding CD0116 13 1.628 1.121 subunit putative oxidoreductase, acetyl-CoA synthase CD0174 4 2.687 1.055 subunit CD0790 putative NUDIX-family hydrolase 8 1.491 1.32 inosine-uridine preferring nucleoside CD1682 iunH 4 0.518 0.984 hydrolase CD2181 putative aromatic compounds hydrolase 7 0372 0.909 CD2342 sucD succinate-semialdehyde dehydrogenase 6 0.651 0.906 Metabolism -

Coordinated Slowing of Metabolism in Enteric Bacteria Under Nitrogen
Coordinated Slowing of Metabolism in Enteric Bacteria under Nitrogen Limitation: A Perspective Ned S. Wingreen NEC Research Institute, 4 Independence Way Princeton, New Jersey 08540 and Department of Physics, University of California Berkeley, CA 94720 Sydney Kustu Department of Plant Biology, Molecular and Cell Biology University of California, Berkeley, CA 94720 Abstract It is natural to ask how bacteria coordinate metabolism when depletion of an essential nutrient limits their growth, and they must slow their entire rate of biosyn- thesis. A major nutrient with a fluctuating abundance is nitrogen. The growth rate of enteric bacteria under nitrogen-limiting conditions is known to correlate with the internal concentration of free glutamine, the glutamine pool. Here we compare the patterns of utilization of L-glutamine and L-glutamate, the two central inter- mediates of nitrogen metabolism. Monomeric precursors of all of the cell’s macro- molecules – proteins, nucleic acids, and surface polymers – require the amide group of glutamine at the first dedicated step of biosynthesis. This is the case even though only a minority (∼12%) of total cell nitrogen derives from glutamine. In contrast, the amino group of glutamate, which provides the remainder of cell nitrogen, is arXiv:physics/0110037v1 [physics.bio-ph] 12 Oct 2001 generally required late in biosynthetic pathways, e.g. in transaminase reactions for amino acid synthesis. We propose that the pattern of glutamine dependence coor- dinates the decrease in biosynthesis under conditions of nitrogen limitation. Hence, the glutamine pool plays a global regulatory role in the cell. 1 INTRODUCTION Enteric bacteria are notable for their varying environment. -

Letters to Nature
letters to nature Received 7 July; accepted 21 September 1998. 26. Tronrud, D. E. Conjugate-direction minimization: an improved method for the re®nement of macromolecules. Acta Crystallogr. A 48, 912±916 (1992). 1. Dalbey, R. E., Lively, M. O., Bron, S. & van Dijl, J. M. The chemistry and enzymology of the type 1 27. Wolfe, P. B., Wickner, W. & Goodman, J. M. Sequence of the leader peptidase gene of Escherichia coli signal peptidases. Protein Sci. 6, 1129±1138 (1997). and the orientation of leader peptidase in the bacterial envelope. J. Biol. Chem. 258, 12073±12080 2. Kuo, D. W. et al. Escherichia coli leader peptidase: production of an active form lacking a requirement (1983). for detergent and development of peptide substrates. Arch. Biochem. Biophys. 303, 274±280 (1993). 28. Kraulis, P.G. Molscript: a program to produce both detailed and schematic plots of protein structures. 3. Tschantz, W. R. et al. Characterization of a soluble, catalytically active form of Escherichia coli leader J. Appl. Crystallogr. 24, 946±950 (1991). peptidase: requirement of detergent or phospholipid for optimal activity. Biochemistry 34, 3935±3941 29. Nicholls, A., Sharp, K. A. & Honig, B. Protein folding and association: insights from the interfacial and (1995). the thermodynamic properties of hydrocarbons. Proteins Struct. Funct. Genet. 11, 281±296 (1991). 4. Allsop, A. E. et al.inAnti-Infectives, Recent Advances in Chemistry and Structure-Activity Relationships 30. Meritt, E. A. & Bacon, D. J. Raster3D: photorealistic molecular graphics. Methods Enzymol. 277, 505± (eds Bently, P. H. & O'Hanlon, P. J.) 61±72 (R. Soc. Chem., Cambridge, 1997). -

Generated by SRI International Pathway Tools Version 25.0, Authors S
Authors: Pallavi Subhraveti Ron Caspi Quang Ong Peter D Karp An online version of this diagram is available at BioCyc.org. Biosynthetic pathways are positioned in the left of the cytoplasm, degradative pathways on the right, and reactions not assigned to any pathway are in the far right of the cytoplasm. Transporters and membrane proteins are shown on the membrane. Ingrid Keseler Periplasmic (where appropriate) and extracellular reactions and proteins may also be shown. Pathways are colored according to their cellular function. Gcf_000725805Cyc: Streptomyces xanthophaeus Cellular Overview Connections between pathways are omitted for legibility. -

Exploring the Chemistry and Evolution of the Isomerases
Exploring the chemistry and evolution of the isomerases Sergio Martínez Cuestaa, Syed Asad Rahmana, and Janet M. Thorntona,1 aEuropean Molecular Biology Laboratory, European Bioinformatics Institute, Wellcome Trust Genome Campus, Hinxton, Cambridge CB10 1SD, United Kingdom Edited by Gregory A. Petsko, Weill Cornell Medical College, New York, NY, and approved January 12, 2016 (received for review May 14, 2015) Isomerization reactions are fundamental in biology, and isomers identifier serves as a bridge between biochemical data and ge- usually differ in their biological role and pharmacological effects. nomic sequences allowing the assignment of enzymatic activity to In this study, we have cataloged the isomerization reactions known genes and proteins in the functional annotation of genomes. to occur in biology using a combination of manual and computa- Isomerases represent one of the six EC classes and are subdivided tional approaches. This method provides a robust basis for compar- into six subclasses, 17 sub-subclasses, and 245 EC numbers cor- A ison and clustering of the reactions into classes. Comparing our responding to around 300 biochemical reactions (Fig. 1 ). results with the Enzyme Commission (EC) classification, the standard Although the catalytic mechanisms of isomerases have already approach to represent enzyme function on the basis of the overall been partially investigated (3, 12, 13), with the flood of new data, an integrated overview of the chemistry of isomerization in bi- chemistry of the catalyzed reaction, expands our understanding of ology is timely. This study combines manual examination of the the biochemistry of isomerization. The grouping of reactions in- chemistry and structures of isomerases with recent developments volving stereoisomerism is straightforward with two distinct types cis-trans in the automatic search and comparison of reactions. -

The Microbiota-Produced N-Formyl Peptide Fmlf Promotes Obesity-Induced Glucose
Page 1 of 230 Diabetes Title: The microbiota-produced N-formyl peptide fMLF promotes obesity-induced glucose intolerance Joshua Wollam1, Matthew Riopel1, Yong-Jiang Xu1,2, Andrew M. F. Johnson1, Jachelle M. Ofrecio1, Wei Ying1, Dalila El Ouarrat1, Luisa S. Chan3, Andrew W. Han3, Nadir A. Mahmood3, Caitlin N. Ryan3, Yun Sok Lee1, Jeramie D. Watrous1,2, Mahendra D. Chordia4, Dongfeng Pan4, Mohit Jain1,2, Jerrold M. Olefsky1 * Affiliations: 1 Division of Endocrinology & Metabolism, Department of Medicine, University of California, San Diego, La Jolla, California, USA. 2 Department of Pharmacology, University of California, San Diego, La Jolla, California, USA. 3 Second Genome, Inc., South San Francisco, California, USA. 4 Department of Radiology and Medical Imaging, University of Virginia, Charlottesville, VA, USA. * Correspondence to: 858-534-2230, [email protected] Word Count: 4749 Figures: 6 Supplemental Figures: 11 Supplemental Tables: 5 1 Diabetes Publish Ahead of Print, published online April 22, 2019 Diabetes Page 2 of 230 ABSTRACT The composition of the gastrointestinal (GI) microbiota and associated metabolites changes dramatically with diet and the development of obesity. Although many correlations have been described, specific mechanistic links between these changes and glucose homeostasis remain to be defined. Here we show that blood and intestinal levels of the microbiota-produced N-formyl peptide, formyl-methionyl-leucyl-phenylalanine (fMLF), are elevated in high fat diet (HFD)- induced obese mice. Genetic or pharmacological inhibition of the N-formyl peptide receptor Fpr1 leads to increased insulin levels and improved glucose tolerance, dependent upon glucagon- like peptide-1 (GLP-1). Obese Fpr1-knockout (Fpr1-KO) mice also display an altered microbiome, exemplifying the dynamic relationship between host metabolism and microbiota. -

K. ENZYME KINETICS and CATALYSIS Basic Kinetics
K. ENZYME KINETICS AND CATALYSIS Biochemical reactions are too slow to support life at room temperature. Whether it is the synthesis of proteins, the metabolism of sugars or the oxidation of fatty acids, all of it proceeds on a time scale unsuitable for life. Moreover, many reactions that are undesirable proceed on a comparable timescale, making it even more difficult to coordinate the necessary chemistry to support life. How useful would it be to have polynucleotides degrading faster than they are synthesized? The basic reactions associated with the synthesis and degradation of essential polymers, and with the metabolism of nutrients all require massive rate accelerations to be of any service to the cell. By specifically accelerating those reactions of interest without accidentally accelerating undesired reactions, as would happen by simply heating the pot, biochemical catalysts shape chemistry in such a way as to permit every biological process required for survival and propagation. Basic Kinetics Reaction Rates The rate of a reaction is defined as the instantaneous rate of change in the concentration of a starting material (“substrate” in biochemical nomenclature – abbreviated “S”) or a product with respect to time. By definition, reaction rates are always positive, which leads to the following definition of reaction rate, abbreviated v (for velocity) in most of the biochemical literature. S → P (eq. K.1) d[P] d[S] v = = − (eq. K.2) dt dt Because these are instantaneous rates of change, they can be obtained as tangents to curves that plot [P] or [S] vs. time. Typically, one takes the initial rate (Figure K.1) – as close to the point of € initiation of the reaction as possible, since the reaction components are well-defined at that moment. -

(12) Patent Application Publication (10) Pub. No.: US 2009/0292100 A1 Fiene Et Al
US 20090292100A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2009/0292100 A1 Fiene et al. (43) Pub. Date: Nov. 26, 2009 (54) PROCESS FOR PREPARING (86). PCT No.: PCT/EP07/57646 PENTAMETHYLENE 1.5-DIISOCYANATE S371 (c)(1), (75) Inventors: Martin Fiene, Niederkirchen (DE): (2), (4) Date: Jan. 9, 2009 (DE);Eckhard Wolfgang Stroefer, Siegel, Mannheim (30) Foreign ApplicationO O Priority Data Limburgerhof (DE); Stephan Aug. 1, 2006 (EP) .................................. O61182.56.4 Freyer, Neustadt (DE); Oskar Zelder, Speyer (DE); Gerhard Publication Classification Schulz, Bad Duerkheim (DE) (51) Int. Cl. Correspondence Address: CSG 18/00 (2006.01) OBLON, SPIVAK, MCCLELLAND MAIER & CD7C 263/2 (2006.01) NEUSTADT, L.L.P. CI2P I3/00 (2006.01) 194O DUKE STREET CD7C 263/10 (2006.01) ALEXANDRIA, VA 22314 (US) (52) U.S. Cl. ........... 528/85; 560/348; 435/128; 560/347; 560/355 (73) Assignee: BASFSE, LUDWIGSHAFEN (DE) (57) ABSTRACT (21) Appl. No.: 12/373,088 The present invention relates to a process for preparing pen tamethylene 1,5-diisocyanate, to pentamethylene 1,5-diiso (22) PCT Filed: Jul. 25, 2007 cyanate prepared in this way and to the use thereof. US 2009/0292100 A1 Nov. 26, 2009 PROCESS FOR PREPARING ene diisocyanates, especially pentamethylene 1,4-diisocyan PENTAMETHYLENE 1.5-DIISOCYANATE ate. Depending on its preparation, this proportion may be up to several % by weight. 0014. The pentamethylene 1,5-diisocyanate prepared in 0001. The present invention relates to a process for pre accordance with the invention has, in contrast, a proportion of paring pentamethylene 1,5-diisocyanate, to pentamethylene the branched pentamethylene diisocyanate isomers of in each 1.5-diisocyanate prepared in this way and to the use thereof. -

Bacterial Selenoproteins: a Role in Pathogenesis and Targets for Antimicrobial Development
University of Central Florida STARS Electronic Theses and Dissertations, 2004-2019 2009 Bacterial Selenoproteins: A Role In Pathogenesis And Targets For Antimicrobial Development Sarah Rosario University of Central Florida Part of the Medical Sciences Commons Find similar works at: https://stars.library.ucf.edu/etd University of Central Florida Libraries http://library.ucf.edu This Doctoral Dissertation (Open Access) is brought to you for free and open access by STARS. It has been accepted for inclusion in Electronic Theses and Dissertations, 2004-2019 by an authorized administrator of STARS. For more information, please contact [email protected]. STARS Citation Rosario, Sarah, "Bacterial Selenoproteins: A Role In Pathogenesis And Targets For Antimicrobial Development" (2009). Electronic Theses and Dissertations, 2004-2019. 3822. https://stars.library.ucf.edu/etd/3822 BACTERIAL SELENOPROTEINS: A ROLE IN PATHOGENESIS AND TARGETS FOR ANTIMICROBIAL DEVELOPMENT. by SARAH E. ROSARIO B.S. Florida State University, 2000 M.P.H. University of South Florida, 2002 A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Burnett School of Biomedical Sciences in the College of Medicine at the University of Central Florida Orlando, Florida Summer Term 2009 Major Professor: William T. Self © 2009 Sarah E. Rosario ii ABSTRACT Selenoproteins are unique proteins in which selenocysteine is inserted into the polypeptide chain by highly specialized translational machinery. They exist within all three kingdoms of life. The functions of these proteins in biology are still being defined. In particular, the importance of selenoproteins in pathogenic microorganisms has received little attention. We first established that a nosocomial pathogen, Clostridium difficile, utilizes a selenoenzyme dependent pathway for energy metabolism. -

Designed Mono- and Di-Covalent Inhibitors Trap Modeled Functional Motions for Trypanosoma Cruzi Proline Racemase in Crystallography
RESEARCH ARTICLE Designed mono- and di-covalent inhibitors trap modeled functional motions for Trypanosoma cruzi proline racemase in crystallography 1¤a 2¤b 2 Patricia de Aguiar Amaral , Delphine Autheman , Guilherme Dias de MeloID , 1 1 2¤c 2¤d Nicolas Gouault , Jean-FrancËois Cupif , Sophie GoyardID , Patricia Dutra , 2 2 3 4 4 Nicolas Coatnoan , Alain Cosson , Damien Monet , Frederick Saul , Ahmed HaouzID , 1 3 2¤e a1111111111 Philippe Uriac *, Arnaud Blondel *, Paola MinoprioID * a1111111111 a1111111111 1 Universite de Rennes 1, Equipe Chimie organique et interfaces (CORINT), UMR 6226 Sciences Chimiques de Rennes, Rennes, France, 2 Institut Pasteur, Laboratoire des Processus Infectieux à TrypanosomatideÂs, a1111111111 DeÂpartement Infection et EpideÂmiologie, Paris, France, 3 Institut Pasteur, Unite de Bioinformatique a1111111111 Structurale, DeÂpartement de Biologie Structurale et Chimie, CNRS-UMR 3528, Paris, France, 4 Institut Pasteur, Plateforme de Cristallographie, DeÂpartement de Biologie Structurale et Chimie, CNRS-UMR 3528, Paris, France ¤a Current address: LaPlaM/PPGCA, Universidade do Extremo Sul Catarinense, CriciuÂmaÐSC, Brazil ¤b Current address: Cell Surface Signalling Laboratory, Wellcome Trust Sanger Institute, Wellcome Trust OPEN ACCESS Genome Campus, Cambridge, United Kingdom Citation: Amaral PdA, Autheman D, de Melo GD, ¤c Current address: Institut Pasteur, Centre d'Innovation et Recherche Technologique, Paris, France Gouault N, Cupif J-F, Goyard S, et al. (2018) ¤d Current address: LaboratoÂrio de BioquõÂmica de ProtozoaÂrios e Imunofisiologia do ExercõÂcio, Departamento de Microbiologia, Imunologia e Parasitologia, Universidade do Estado do Rio de Janeiro, Rio Designed mono- and di-covalent inhibitors trap de Janeiro, Brazil modeled functional motions for Trypanosoma cruzi ¤e Current address: Trypanosomatids Infectious Processes Laboratory, Scientific Platform Pasteur-USP, proline racemase in crystallography. -

Discovery of a Novel Amino Acid Racemase Through Exploration of Natural Variation in Arabidopsis Thaliana
Discovery of a novel amino acid racemase through exploration of natural variation in Arabidopsis thaliana Renee C. Straucha,b, Elisabeth Svedinc, Brian Dilkesc, Clint Chappled, and Xu Lia,b,1 aPlants for Human Health Institute, North Carolina State University, Kannapolis, NC 28081; bDepartment of Plant and Microbial Biology, North Carolina State University, Raleigh, NC 27695; cDepartment of Horticulture and Landscape Architecture, Purdue University, West Lafayette, IN 47907; and dDepartment of Biochemistry, Purdue University, West Lafayette, IN 47907 Edited by Justin O. Borevitz, Australian National University, Canberra, ACT, Australia, and accepted by the Editorial Board August 1, 2015 (received for review February 16, 2015) Plants produce diverse low-molecular-weight compounds via spe- contribution to the variation in at least three-fourths of detected cialized metabolism. Discovery of the pathways underlying produc- mass peaks (8). tion of these metabolites is an important challenge for harnessing Here we describe an integrated transdisciplinary platform, the huge chemical diversity and catalytic potential in the plant king- combining metabolomics, genetics, and genomics, to exploit the dom for human uses, but this effort is often encumbered by the biochemical and genetic diversity of natural accessions of the necessity to initially identify compounds of interest or purify a cata- model plant A. thaliana to uncover associations between genes lyst involved in their synthesis. As an alternative approach, we have and metabolites. Using this platform, we linked a differentially performed untargeted metabolite profiling and genome-wide asso- accumulating metabolite, identified through chemical analysis as ciation analysis on 440 natural accessions of Arabidopsis thaliana. N-malonyl-D-allo-isoleucine (NMD-Ile), to a previously unchar- This approach allowed us to establish genetic linkages between acterized gene identified as an amino acid racemase through metabolites and genes.