Proximal Pathway Enrichment Analysis for Targeting Comorbid Diseases Via Network Endopharmacology
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
Use of Intravenous Esmolol to Predict Efficacy of Oral Beta-Adrenergic Blocker Therapy in Patients with Neurocardiogenic Syncope
402 JACC Vol. 19, No. 2 February 1992:402-8 REPORTS ON THERAPY Use of Intravenous Esmolol to Predict Efficacy of Oral Beta Adrenergic Blocker Therapy in Patients With Neurocardiogenic Syncope JASBIR S. SRA, MD, VISHNUBHAKTA S. MURTHY, MD, PHD, MOHAMMAD R. JAZAYERI, MD, FACC, YUE-HUA SHEN, MD, PAUL J. TROUP, MD, FACC, BOAZ AVITALL, MD, PHD, MASOOD AKHTAR, MD, FACC Milwaukee, Wisconsin The usefulness of esmolol in predicting the efficacy of treatment Irrespective of their response to esmolol infusion, all patients with an oral beta-adrenergic blocking agent was evaluated in 27 had a follow-up tilt test with oral metoprolol after an interval of consecutive patients with neurocardiogenic syncope. Seventeen ~5 half-lives of the drug. All 16 patients (100%) with a negative patients had a positive head-up tilt test response at baseline and 10 tilt test response during esmolol infusion had a negative tilt test patients required intravenous isoproterenol for provocation of response with oral metoprolol. Of the 11 patients with a positive hypotension. All patients were then given a continuous esmolol tilt test response during esmolol infusion, 10 (90%) continued to infusion (500 JLg/kg per min loading dose for 3 min followed by have a positive response with oral metoprolol. 300 Jlg/kg per min maintenance dose) and rechallenged with a It is concluded that in the electrophysiology laboratory, es head-up tilt test at baseline or with isoproterenol. molol can accurately predict the outcome of a head-up tilt Of the 17 patients with a positive baseline tilt test response, 11 response to oral metoprolol. -
Esmolol Hydrochloride 10 Mg/Ml Solution for Injection Esmolol Hydrochloride
Package leaflet: Information for the patient Esmolol hydrochloride 10 mg/ml solution for injection Esmolol hydrochloride Read all of this leaflet carefully before you start using this medicine because it contains important information for you. – Keep this leaflet. You may need to read it again. – If you have any further questions, ask your doctor or nurse. – This medicine has been prescribed for you. Do not pass it on to others. It may harm them, even if their symptoms are the same as yours. – If you get any side effects, talk to your doctor. This includes any possible side effects not listed in this leaflet. See section 4. What is in this leaflet: 1. What Esmolol hydrochloride 10 mg/ml solution for injection is and what it is used for 2. What you need to know before you use Esmolol hydrochloride 10 mg/ml solution for injection 3. How to use Esmolol hydrochloride 10 mg/ml solution for injection 4. Possible side effects 5. How to store Esmolol hydrochloride 10 mg/ml solution for injection 6. Contents of the pack and other information 1. What Esmolol hydrochloride 10 mg/ml chlor promazine, used to treat mental dis - solution for injection is and what it is orders) used for – Clozapine which is used to treat mental Esmolol hydrochloride 10 mg/ml belongs to the disorders group of beta-blockers. These medicines slow – Epinephrine, which is used to treat allergic down the heartbeat and reduce blood pressure. reactions – Medicines used to treat asthma Esmolol hydrochloride 10 mg/ml is used for – Medicines used to treat colds or a blocked short-term treatment if your heart beats too nose, called nasal “decongestants” fast. -

Crohn's & Colitis Program, Patient Information Guide
Inflammatory Bowel Disease Program Patient Information Guide Page # Contents ..........................................................................................................................................1 Welcome! Welcome Letter ....................................................................................................................3 Important Things to Know Up Front ....................................................................................4 Meet Your Crohn’s & Colitis Team .....................................................................................6 How to Contact Us Crohn’s & Colitis Clinic Schedule .....................................................................................11 Physician and Nurse Contact Information..........................................................................12 Appointment Scheduling and Other Contact Information..................................................13 The Basics of Inflammatory Bowel Disease (IBD) Basic Information about Inflammatory Bowel Disease (IBD) ...........................................14 Frequently Asked Questions about Inflammatory Bowel Disease (IBD) ..........................18 Testing in IBD Colonoscopy and Flexible Sigmoidoscopy ........................................................................20 Upper Endoscopy ...............................................................................................................21 Capsule Endoscopy and Deep Enteroscopy .......................................................................22 -

Inflammatory Bowel Disease Agents
Therapeutic Class Overview Inflammatory Bowel Disease Agents INTRODUCTION • Inflammatory bowel disease (IBD) is a spectrum of chronic idiopathic inflammatory intestinal conditions that cause gastrointestinal symptoms including diarrhea, abdominal pain, bleeding, fatigue, and weight loss. The exact cause of IBD is unknown; however, proposed etiologies involve a combination of infectious, genetic, and lifestyle factors (Bernstein et al 2015, Peppercorn 2019[a], Peppercorn 2020[c]). • Complications of IBD include hemorrhage, rectal fissures, fistulas, peri-rectal and intra-abdominal abscesses, and colon cancer. Possible extra-intestinal complications include hepatobiliary complications, anemia, arthritis and arthralgias, uveitis, skin lesions, and mood and anxiety disorders (Bernstein et al 2015). • Ulcerative colitis (UC) and Crohn’s disease (CD) are 2 forms of IBD that differ in pathophysiology and presentation; as a result of these differences, the approach to the treatment of each condition often differs (Peppercorn 2019[a]). • UC is characterized by recurrent episodes of inflammation of the mucosal layer of the colon. The inflammation, limited to the mucosa, commonly involves the rectum and may extend in a proximal and continuous fashion to affect other parts of the colon. The hallmark clinical symptom is an inflamed rectum with symptoms of urgency, bleeding, and tenesmus (Peppercorn 2020[c], Rubin et al 2019). • CD can involve any part of the gastrointestinal tract and is characterized by transmural inflammation and “skip areas.” Transmural inflammation may lead to fibrosis, strictures, sinus tracts, and fistulae (Peppercorn 2019[b]). • The immune system is known to play a critical role in the underlying pathogenesis of IBD. It is suggested that abnormal responses of both innate and adaptive immunity mechanisms induce aberrant intestinal tract inflammation in IBD patients (Geremia et al 2014). -

New Brunswick Drug Plans Formulary
New Brunswick Drug Plans Formulary August 2019 Administered by Medavie Blue Cross on Behalf of the Government of New Brunswick TABLE OF CONTENTS Page Introduction.............................................................................................................................................I New Brunswick Drug Plans....................................................................................................................II Exclusions............................................................................................................................................IV Legend..................................................................................................................................................V Anatomical Therapeutic Chemical (ATC) Classification of Drugs A Alimentary Tract and Metabolism 1 B Blood and Blood Forming Organs 23 C Cardiovascular System 31 D Dermatologicals 81 G Genito Urinary System and Sex Hormones 89 H Systemic Hormonal Preparations excluding Sex Hormones 100 J Antiinfectives for Systemic Use 107 L Antineoplastic and Immunomodulating Agents 129 M Musculo-Skeletal System 147 N Nervous System 156 P Antiparasitic Products, Insecticides and Repellants 223 R Respiratory System 225 S Sensory Organs 234 V Various 240 Appendices I-A Abbreviations of Dosage forms.....................................................................A - 1 I-B Abbreviations of Routes................................................................................A - 4 I-C Abbreviations of Units...................................................................................A -

Properties and Units in Clinical Pharmacology and Toxicology
Pure Appl. Chem., Vol. 72, No. 3, pp. 479–552, 2000. © 2000 IUPAC INTERNATIONAL FEDERATION OF CLINICAL CHEMISTRY AND LABORATORY MEDICINE SCIENTIFIC DIVISION COMMITTEE ON NOMENCLATURE, PROPERTIES, AND UNITS (C-NPU)# and INTERNATIONAL UNION OF PURE AND APPLIED CHEMISTRY CHEMISTRY AND HUMAN HEALTH DIVISION CLINICAL CHEMISTRY SECTION COMMISSION ON NOMENCLATURE, PROPERTIES, AND UNITS (C-NPU)§ PROPERTIES AND UNITS IN THE CLINICAL LABORATORY SCIENCES PART XII. PROPERTIES AND UNITS IN CLINICAL PHARMACOLOGY AND TOXICOLOGY (Technical Report) (IFCC–IUPAC 1999) Prepared for publication by HENRIK OLESEN1, DAVID COWAN2, RAFAEL DE LA TORRE3 , IVAN BRUUNSHUUS1, MORTEN ROHDE1, and DESMOND KENNY4 1Office of Laboratory Informatics, Copenhagen University Hospital (Rigshospitalet), Copenhagen, Denmark; 2Drug Control Centre, London University, King’s College, London, UK; 3IMIM, Dr. Aiguader 80, Barcelona, Spain; 4Dept. of Clinical Biochemistry, Our Lady’s Hospital for Sick Children, Crumlin, Dublin 12, Ireland #§The combined Memberships of the Committee and the Commission (C-NPU) during the preparation of this report (1994–1996) were as follows: Chairman: H. Olesen (Denmark, 1989–1995); D. Kenny (Ireland, 1996); Members: X. Fuentes-Arderiu (Spain, 1991–1997); J. G. Hill (Canada, 1987–1997); D. Kenny (Ireland, 1994–1997); H. Olesen (Denmark, 1985–1995); P. L. Storring (UK, 1989–1995); P. Soares de Araujo (Brazil, 1994–1997); R. Dybkær (Denmark, 1996–1997); C. McDonald (USA, 1996–1997). Please forward comments to: H. Olesen, Office of Laboratory Informatics 76-6-1, Copenhagen University Hospital (Rigshospitalet), 9 Blegdamsvej, DK-2100 Copenhagen, Denmark. E-mail: [email protected] Republication or reproduction of this report or its storage and/or dissemination by electronic means is permitted without the need for formal IUPAC permission on condition that an acknowledgment, with full reference to the source, along with use of the copyright symbol ©, the name IUPAC, and the year of publication, are prominently visible. -

Supplementary Information
Supplementary Information Network-based Drug Repurposing for Novel Coronavirus 2019-nCoV Yadi Zhou1,#, Yuan Hou1,#, Jiayu Shen1, Yin Huang1, William Martin1, Feixiong Cheng1-3,* 1Genomic Medicine Institute, Lerner Research Institute, Cleveland Clinic, Cleveland, OH 44195, USA 2Department of Molecular Medicine, Cleveland Clinic Lerner College of Medicine, Case Western Reserve University, Cleveland, OH 44195, USA 3Case Comprehensive Cancer Center, Case Western Reserve University School of Medicine, Cleveland, OH 44106, USA #Equal contribution *Correspondence to: Feixiong Cheng, PhD Lerner Research Institute Cleveland Clinic Tel: +1-216-444-7654; Fax: +1-216-636-0009 Email: [email protected] Supplementary Table S1. Genome information of 15 coronaviruses used for phylogenetic analyses. Supplementary Table S2. Protein sequence identities across 5 protein regions in 15 coronaviruses. Supplementary Table S3. HCoV-associated host proteins with references. Supplementary Table S4. Repurposable drugs predicted by network-based approaches. Supplementary Table S5. Network proximity results for 2,938 drugs against pan-human coronavirus (CoV) and individual CoVs. Supplementary Table S6. Network-predicted drug combinations for all the drug pairs from the top 16 high-confidence repurposable drugs. 1 Supplementary Table S1. Genome information of 15 coronaviruses used for phylogenetic analyses. GenBank ID Coronavirus Identity % Host Location discovered MN908947 2019-nCoV[Wuhan-Hu-1] 100 Human China MN938384 2019-nCoV[HKU-SZ-002a] 99.99 Human China MN975262 -

Prediction of Premature Termination Codon Suppressing Compounds for Treatment of Duchenne Muscular Dystrophy Using Machine Learning
Prediction of Premature Termination Codon Suppressing Compounds for Treatment of Duchenne Muscular Dystrophy using Machine Learning Kate Wang et al. Supplemental Table S1. Drugs selected by Pharmacophore-based, ML-based and DL- based search in the FDA-approved drugs database Pharmacophore WEKA TF 1-Palmitoyl-2-oleoyl-sn-glycero-3- 5-O-phosphono-alpha-D- (phospho-rac-(1-glycerol)) ribofuranosyl diphosphate Acarbose Amikacin Acetylcarnitine Acetarsol Arbutamine Acetylcholine Adenosine Aldehydo-N-Acetyl-D- Benserazide Acyclovir Glucosamine Bisoprolol Adefovir dipivoxil Alendronic acid Brivudine Alfentanil Alginic acid Cefamandole Alitretinoin alpha-Arbutin Cefdinir Azithromycin Amikacin Cefixime Balsalazide Amiloride Cefonicid Bethanechol Arbutin Ceforanide Bicalutamide Ascorbic acid calcium salt Cefotetan Calcium glubionate Auranofin Ceftibuten Cangrelor Azacitidine Ceftolozane Capecitabine Benserazide Cerivastatin Carbamoylcholine Besifloxacin Chlortetracycline Carisoprodol beta-L-fructofuranose Cilastatin Chlorobutanol Bictegravir Citicoline Cidofovir Bismuth subgallate Cladribine Clodronic acid Bleomycin Clarithromycin Colistimethate Bortezomib Clindamycin Cyclandelate Bromotheophylline Clofarabine Dexpanthenol Calcium threonate Cromoglicic acid Edoxudine Capecitabine Demeclocycline Elbasvir Capreomycin Diaminopropanol tetraacetic acid Erdosteine Carbidopa Diazolidinylurea Ethchlorvynol Carbocisteine Dibekacin Ethinamate Carboplatin Dinoprostone Famotidine Cefotetan Dipyridamole Fidaxomicin Chlormerodrin Doripenem Flavin adenine dinucleotide -

33-Adrenoceptor-Mediated Relaxation Induced by Isoprenaline And
J. Smooth Muscle Res. 33 : 99-106. 99 The )32 and [33-Adrenoceptor-Mediated Relaxation Induced by Isoprenaline and Salbutamol in Guinea Pig Taenia Caecum Katsuo KOIKE, Tsukasa IcHiNo, Takahiro HORINOUCHI and Issei TAKAYANAGI Departmentof Chemical Pharmacology, Toho University School of PharmaceuticalSciences, 2-2-1, Miyama,Funabashi, Chiba 274, Japan Abstract To understand the receptor subtypes responsible for /3adrenoceptormediated relaxa tion of guinea pig taenia caecum, we investigated the effects of isoprenaline and salbutamol . Isoprenaline and salbutamol caused dose-dependent relaxation of the guinea pig taenia caecum. Propranolol, bupranolol and butoxamine produced shifts of the concentration response curves for isoprenaline and salbutamol. Schild regression analyses carried out for propranolol against isoprenaline and salbutamol gave pA2 values of 8.43 and 8.88, respective ly. Schild regression analyses carried out for butoxamine against isoprenaline and sal butamol gave pA2 values of 6.46 and 6.68, respectively. Schild regression analyses carried out for bupranolol against isoprenaline and salbutamol gave pA2 values of 8.60 and 8.69, respectively. However, in the presence of 3 x 10' M atenolol, 10-4 M butoxamine and 10-6 M phentolamine to block the fir , /32 and a -adrenoceptor effects, respectively, Schild regression analyses carried out for bupranolol against isoprenaline and salbutamol gave pA2 values of 5.77 and 5.97, respectively. These results suggest that the relaxant responses to isoprenaline and salbutamol in the guinea pig taenia caecum are mediated by both the /.32 and the A-adrenoceptors. Key words : f2-adrenoceptor, A-adrenoceptor, isoprenaline, salbutamol , guinea pig taenia caecum Introduction The /3adrenoceptors were subclassified as and /32subtypes based on the agonist potency and tissue localization. -

What Is the Extent of the Role Played by Genetic Factors in Periodontitis?
Perio Insight 4 Summer 2017 www.efp.org/perioinsight DEBATE EXPERT VIEW FOCUS RESEARCH Editor: Joanna Kamma What is the extent of the role played by genetic factors in periodontitis? Discover latest JCP research hile genetic factors are known to play a role in In a comprehensive overview of current knowledge, The Journal of Clinical Periodontology Wperiodontitis, a lot of research still needs to be they outline what is known today and discuss the (JCP) is the official scientific publication done to understand the mechanisms that are involved. most promising lines for future inquiry. of the European Federation of Indeed, the limited extent to which the genetic They note that the phenotypes of aggressive Periodontology (EFP). It publishes factors associated with periodontitis have been periodontitis (AgP) and chronic periodontitis (CP) may research relating to periodontal and peri- identified is “somewhat disappointing”, say Bruno not be as distinct as previously assumed because they implant diseases and their treatment. G. Loos and Deon P.M. Chin, from the Academic share genetic and other risk factors. The six JCP articles summarised in this Centre for Dentistry in Amsterdam. More on pages 2-5 edition of Perio Insight cover: (1) how periodontitis changes renal structures by oxidative stress and lipid peroxidation; (2) gingivitis and lifestyle influences on high-sensitivity C-reactive protein and interleukin 6 in adolescents; (3) the long-term efficacy of periodontal Researchers call regenerative therapies; (4) tooth loss in generalised aggressive periodontitis; (5) the association between diabetes for global action mellitus/hyperglycaemia and peri- implant diseases; (6) the efficacy of on periodontal collagen matrix seal and collagen sponge on ridge preservation in disease combination with bone allograft. -
Guidance for the Format and Content of the Protocol of Non-Interventional
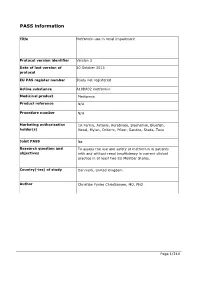
PASS information Title Metformin use in renal impairment Protocol version identifier Version 2 Date of last version of 30 October 2013 protocol EU PAS register number Study not registered Active substance A10BA02 metformin Medicinal product Metformin Product reference N/A Procedure number N/A Marketing authorisation 1A Farma, Actavis, Aurobindo, Biochemie, Bluefish, holder(s) Hexal, Mylan, Orifarm, Pfizer, Sandoz, Stada, Teva Joint PASS No Research question and To assess the use and safety of metformin in patients objectives with and without renal insufficiency in current clinical practice in at least two EU Member States. Country(-ies) of study Denmark, United Kingdom Author Christian Fynbo Christiansen, MD, PhD Page 1/214 Marketing authorisation holder(s) Marketing authorisation N/A holder(s) MAH contact person N/A Page 2/214 1. Table of Contents PASS information .......................................................................................................... 1 Marketing authorisation holder(s) .................................................................................... 2 1. Table of Contents ...................................................................................................... 3 2. List of abbreviations ................................................................................................... 4 3. Responsible parties .................................................................................................... 5 4. Abstract .................................................................................................................. -

OCALIVA™ (Obeticholic Acid) Oral
PHARMACY COVERAGE GUIDELINES ORIGINAL EFFECTIVE DATE: 9/15/2016 SECTION: DRUGS LAST REVIEW DATE: 8/19/2021 LAST CRITERIA REVISION DATE: 8/19/2021 ARCHIVE DATE: OCALIVA™ (obeticholic acid) oral Coverage for services, procedures, medical devices and drugs are dependent upon benefit eligibility as outlined in the member's specific benefit plan. This Pharmacy Coverage Guideline must be read in its entirety to determine coverage eligibility, if any. This Pharmacy Coverage Guideline provides information related to coverage determinations only and does not imply that a service or treatment is clinically appropriate or inappropriate. The provider and the member are responsible for all decisions regarding the appropriateness of care. Providers should provide BCBSAZ complete medical rationale when requesting any exceptions to these guidelines. The section identified as “Description” defines or describes a service, procedure, medical device or drug and is in no way intended as a statement of medical necessity and/or coverage. The section identified as “Criteria” defines criteria to determine whether a service, procedure, medical device or drug is considered medically necessary or experimental or investigational. State or federal mandates, e.g., FEP program, may dictate that any drug, device or biological product approved by the U.S. Food and Drug Administration (FDA) may not be considered experimental or investigational and thus the drug, device or biological product may be assessed only on the basis of medical necessity. Pharmacy Coverage Guidelines are subject to change as new information becomes available. For purposes of this Pharmacy Coverage Guideline, the terms "experimental" and "investigational" are considered to be interchangeable. BLUE CROSS®, BLUE SHIELD® and the Cross and Shield Symbols are registered service marks of the Blue Cross and Blue Shield Association, an association of independent Blue Cross and Blue Shield Plans.