Jemds.Com Case Report
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Tracheal Perforation in a Neonate: a Devastating
Letters to Editor Tracheal perforation in a size of endotracheal tube, cuff overinflation, cough and vigorous movements of head and neck.[2,3] neonate: A devastating Neonatal tracheal injury is ascribed to factors like complication following traumatic traumatic delivery, weak trachea, congenital tracheal endotracheal intubation stenosis, ring agenesis, metal stylets, rigid endotracheal tube, excessive external laryngeal pressure and [1,4,5] Sir, prolonged ventilation. Tracheal injury in neonates following endotracheal In our case, rigorous attempts at intubation along with intubation represents an uncommon complication excessive hyperextension of head and neck due to altered rarely described in literature but carries high anatomy are likely to have contributed to tracheal injury. morbidity and a mortality rate of 70%.[1] We describe Multiple attempts at intubation are known to result in a [4] a case of neonatal tracheal perforation following false tract and are related to anterior tracheal lesions. multiple attempts at endotracheal intubation due to an Incidentally, our case was an undiagnosed Pierre unanticipated difficulty in an emergency situation in Robins Sequence. A small receding mandible, tongue an undiagnosed case of Pierre Robin Sequence. immobility and cleft palate are identified as independent factors for difficult airway with a risk of upper airway A 4-week-old female baby presented to the emergency obstruction, difficult mask holding, difficult intubation, department with respiratory difficulty. In view of severe leak through the cleft resulting in inadequate ventilation respiratory distress and desaturation (SpO2- 60%) a as well as passage of the endotracheal tube into the decision was made to provide ventilatory support to cleft.[6] Successful airway management in such situation the neonate. -

Challenges in Long-Term Control of Hypercalcaemia with Denosumab
Taylor-Miller, T., Sivaprakasam, P., Smithson, S. F., Steward, C. G., & Burren, C. P. (2021). Challenges in long-term control of hypercalcaemia with denosumab after haematopoietic stem cell transplantation for TNFRSF11A osteoclast-poor autosomal recessive osteopetrosis. Bone reports, 14, 100738. [100738]. https://doi.org/10.1016/j.bonr.2020.100738 Publisher's PDF, also known as Version of record License (if available): CC BY-NC-ND Link to published version (if available): 10.1016/j.bonr.2020.100738 Link to publication record in Explore Bristol Research PDF-document This is the final published version of the article (version of record). It first appeared online via Elsevier at https://doi.org/10.1016/j.bonr.2020.100738. Please refer to any applicable terms of use of the publisher. University of Bristol - Explore Bristol Research General rights This document is made available in accordance with publisher policies. Please cite only the published version using the reference above. Full terms of use are available: http://www.bristol.ac.uk/red/research-policy/pure/user-guides/ebr-terms/ Bone Reports 14 (2021) 100738 Contents lists available at ScienceDirect Bone Reports journal homepage: www.elsevier.com/locate/bonr Case Report Challenges in long-term control of hypercalcaemia with denosumab after haematopoietic stem cell transplantation for TNFRSF11A osteoclast-poor autosomal recessive osteopetrosis Tashunka Taylor-Miller a, Ponni Sivaprakasam b, Sarah F. Smithson c,d, Colin G. Steward d,e, Christine P. Burren a,d,* a Department of Paediatric -
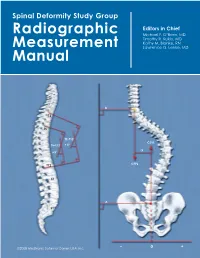
Spinal Deformity Study Group
Spinal Deformity Study Group Editors in Chief Radiographic Michael F. O’Brien, MD Timothy R. Kuklo, MD Kathy M. Blanke, RN Measurement Lawrence G. Lenke, MD Manual B T2 T5 T2–T12 CSVL T5–T12 +X° -X +X° C7PL T12 L2 A S1 ©2008 Medtronic Sofamor Danek USA, Inc. – 0 + Radiographic Measurement Manual Editors in Chief Michael F. O’Brien, MD Timothy R. Kuklo, MD Kathy M. Blanke, RN Lawrence G. Lenke, MD Section Editors Keith H. Bridwell, MD Kathy M. Blanke, RN Christopher L. Hamill, MD William C. Horton, MD Timothy R. Kuklo, MD Hubert B. Labelle, MD Lawrence G. Lenke, MD Michael F. O’Brien, MD David W. Polly Jr, MD B. Stephens Richards III, MD Pierre Roussouly, MD James O. Sanders, MD ©2008 Medtronic Sofamor Danek USA, Inc. Acknowledgements Radiographic Measurement Manual The radiographic measurement manual has been developed to present standardized techniques for radiographic measurement. In addition, this manual will serve as a complimentary guide for the Spinal Deformity Study Group’s radiographic measurement software. Special thanks to the following members of the Spinal Deformity Study Group in the development of this manual. Sigurd Berven, MD Hubert B. Labelle, MD Randal Betz, MD Lawrence G. Lenke, MD Fabien D. Bitan, MD Thomas G. Lowe, MD John T. Braun, MD John P. Lubicky, MD Keith H. Bridwell, MD Steven M. Mardjetko, MD Courtney W. Brown, MD Richard E. McCarthy, MD Daniel H. Chopin, MD Andrew A. Merola, MD Edgar G. Dawson, MD Michael Neuwirth, MD Christopher DeWald, MD Peter O. Newton, MD Mohammad Diab, MD Michael F. -

[email protected] Patient Information Patient Name (Last, First, Middle) Birth Date (Mm-Dd-Yyyy) Sex Male Female
Marfan and Related Disorders Patient Information Instructions: The accurate interpretation and reporting of the genetic results is contingent upon the reason for referral, clinical information, ethnic background, and family history. To help provide the best possible service, supply the information requested below and send paperwork with the specimen, or return by fax to Mayo Clinic Laboratories, Attn: Personalized Genomics Laboratory Genetic Counselors at 507-284-1759. Phone: 507-266-5700 / International clients: +1-507-266-5700 or email [email protected] Patient Information Patient Name (Last, First, Middle) Birth Date (mm-dd-yyyy) Sex Male Female Referring Provider Name (Last, First) Phone Fax* Other Contact Name (Last, First) Phone Fax* *Fax number given must be from a fax machine that complies with applicable HIPAA regulations. Is this a postmortem specimen? Yes No If yes, attach autopsy report if available. Clinical History Reason for Testing (Check all that apply.) Diagnosis Carrier testing Presymptomatic diagnosis Family history Sudden death Note: Genetic testing should always be initiated on an affected family member first, if possible, in order to be most informative for at-risk relatives. See Ethnic Background and Family History section for more information. Diagnosis/Suspected Diagnosis Marfan Syndrome Ehlers-Danlos Syndrome Loeys-Dietz Syndrome Familial thoracic aortic aneurysm and dissection Other: _______________________________________________________________________________________________ Indicate whether the following -

Craniosynostosis and Related Disorders Mark S
CLINICAL REPORT Guidance for the Clinician in Rendering Pediatric Care Identifying the Misshapen Head: Craniosynostosis and Related Disorders Mark S. Dias, MD, FAAP, FAANS,a Thomas Samson, MD, FAAP,b Elias B. Rizk, MD, FAAP, FAANS,a Lance S. Governale, MD, FAAP, FAANS,c Joan T. Richtsmeier, PhD,d SECTION ON NEUROLOGIC SURGERY, SECTION ON PLASTIC AND RECONSTRUCTIVE SURGERY Pediatric care providers, pediatricians, pediatric subspecialty physicians, and abstract other health care providers should be able to recognize children with abnormal head shapes that occur as a result of both synostotic and aSection of Pediatric Neurosurgery, Department of Neurosurgery and deformational processes. The purpose of this clinical report is to review the bDivision of Plastic Surgery, Department of Surgery, College of characteristic head shape changes, as well as secondary craniofacial Medicine and dDepartment of Anthropology, College of the Liberal Arts characteristics, that occur in the setting of the various primary and Huck Institutes of the Life Sciences, Pennsylvania State University, State College, Pennsylvania; and cLillian S. Wells Department of craniosynostoses and deformations. As an introduction, the physiology and Neurosurgery, College of Medicine, University of Florida, Gainesville, genetics of skull growth as well as the pathophysiology underlying Florida craniosynostosis are reviewed. This is followed by a description of each type of Clinical reports from the American Academy of Pediatrics benefit from primary craniosynostosis (metopic, unicoronal, bicoronal, sagittal, lambdoid, expertise and resources of liaisons and internal (AAP) and external reviewers. However, clinical reports from the American Academy of and frontosphenoidal) and their resultant head shape changes, with an Pediatrics may not reflect the views of the liaisons or the emphasis on differentiating conditions that require surgical correction from organizations or government agencies that they represent. -

Spondylolysis and Spondylolisthesis in Children and Adolescents: I
Spondylolysis and Spondylolisthesis in Children and Adolescents: I. Diagnosis, Natural History, and Nonsurgical Management Ralph Cavalier, MD Abstract Martin J. Herman, MD Spondylolysis and spondylolisthesis are often diagnosed in children Emilie V. Cheung, MD presenting with low back pain. Spondylolysis refers to a defect of Peter D. Pizzutillo, MD the vertebral pars interarticularis. Spondylolisthesis is the forward translation of one vertebral segment over the one beneath it. Isth- Dr. Cavalier is Attending Orthopaedic mic spondylolysis, isthmic spondylolisthesis, and stress reactions Surgeon, Summit Sports Medicine and involving the pars interarticularis are the most common forms seen Orthopaedic Surgery, Brunswick, GA. Dr. Herman is Associate Professor, in children. Typical presentation is characterized by a history of Department of Orthopaedic Surgery, activity-related low back pain and the presence of painful spinal Drexel University College of Medicine, mobility and hamstring tightness without radiculopathy. Plain ra- St. Christopher’s Hospital for Children, Philadelphia, PA. Dr. Cheung is Fellow, diography, computed tomography, and single-photon emission Department of Orthopaedic Surgery, computed tomography are useful for establishing the diagnosis. Mayo Clinic, Rochester, MN. Dr. Symptomatic stress reactions of the pars interarticularis or adjacent Pizzutillo is Professor, Department of vertebral structures are best treated with immobilization of the Orthopaedic Surgery, Drexel University College of Medicine, St. Christopher’s spine and activity restriction. Spondylolysis often responds to brief Hospital for Children. periods of activity restriction, immobilization, and physiotherapy. None of the following authors or the Low-grade spondylolisthesis (≤50% translation) is treated similarly. departments with which they are The less common dysplastic spondylolisthesis with intact posterior affiliated has received anything of value elements requires greater caution. -

Failure to Maintain Segmental Lordosis During TLIF for One-Level
European Spine Journal (2019) 28:745–750 https://doi.org/10.1007/s00586-019-05890-w ORIGINAL ARTICLE Failure to maintain segmental lordosis during TLIF for one‑level degenerative spondylolisthesis negatively afects clinical outcome 5 years postoperatively: a prospective cohort of 57 patients Matevž Kuhta1 · Klemen Bošnjak2 · Rok Vengust2 Received: 14 June 2018 / Revised: 28 November 2018 / Accepted: 13 January 2019 / Published online: 24 January 2019 © Springer-Verlag GmbH Germany, part of Springer Nature 2019 Abstract Purpose The present study aimed to determine whether obtaining adequate lumbar (LL) or segmental (SL) lordosis during instrumented TLIF for one-level degenerative spondylolisthesis afects midterm clinical outcome. Methods The study was designed as a prospective one, including 57 patients who underwent single-level TLIF surgery for degenerative spondylolisthesis. Patients were analyzed globally with additional subgroup analysis according to pelvic incidence (PI). Radiographic analysis of spinopelvic sagittal parameters was conducted pre- and postoperatively. Clinical examination including ODI score was performed preoperatively, 1 and 5 years postoperatively. Results Signifcant improvement in ODI scores at 1 and 5 years postoperatively (p < 0.001) was demonstrated. There was a signifcant correlation between anterior shift of SVA and failure to improve SL (p = 0.046). Moreover, anterior SVA shift correlated with increased values of ODI score both 1 and 5 years postoperatively. In low-PI group, failure to correct LL correlated with high ODI scores 5 years postoperatively (r = − 0.499, p = 0.005). Conclusions Failure to correct segmental lordosis during surgery for one-level degenerative spondylolisthesis resulted in anterior displacement of the center of gravity, which in turn correlated with unfavorable clinical outcome 1 and 5 years postoperatively. -

Blueprint Genetics Craniosynostosis Panel
Craniosynostosis Panel Test code: MA2901 Is a 38 gene panel that includes assessment of non-coding variants. Is ideal for patients with craniosynostosis. About Craniosynostosis Craniosynostosis is defined as the premature fusion of one or more cranial sutures leading to secondary distortion of skull shape. It may result from a primary defect of ossification (primary craniosynostosis) or, more commonly, from a failure of brain growth (secondary craniosynostosis). Premature closure of the sutures (fibrous joints) causes the pressure inside of the head to increase and the skull or facial bones to change from a normal, symmetrical appearance resulting in skull deformities with a variable presentation. Craniosynostosis may occur in an isolated setting or as part of a syndrome with a variety of inheritance patterns and reccurrence risks. Craniosynostosis occurs in 1/2,200 live births. Availability 4 weeks Gene Set Description Genes in the Craniosynostosis Panel and their clinical significance Gene Associated phenotypes Inheritance ClinVar HGMD ALPL Odontohypophosphatasia, Hypophosphatasia perinatal lethal, AD/AR 78 291 infantile, juvenile and adult forms ALX3 Frontonasal dysplasia type 1 AR 8 8 ALX4 Frontonasal dysplasia type 2, Parietal foramina AD/AR 15 24 BMP4 Microphthalmia, syndromic, Orofacial cleft AD 8 39 CDC45 Meier-Gorlin syndrome 7 AR 10 19 EDNRB Hirschsprung disease, ABCD syndrome, Waardenburg syndrome AD/AR 12 66 EFNB1 Craniofrontonasal dysplasia XL 28 116 ERF Craniosynostosis 4 AD 17 16 ESCO2 SC phocomelia syndrome, Roberts syndrome -

Cervical Sagittal Alignment in Adolescent High Dysplastic
Guo et al. Journal of Orthopaedic Surgery and Research (2020) 15:243 https://doi.org/10.1186/s13018-020-01762-y RESEARCH ARTICLE Open Access Cervical sagittal alignment in adolescent high dysplastic developmental spondylolisthesis: how does the cervical spine respond to the reduction of spondylolisthesis? Xinhu Guo, Weishi Li* , Zhongqiang Chen, Zhaoqing Guo, Qiang Qi, Yan Zeng, Chuiguo Sun and Woquan Zhong Abstract Background: Although pelvic and related parameters have been well stated in lumbar developmental spondylolisthesis, cervical sagittal alignment in these patients is poorly studied, especially in high dysplastic developmental spondylolisthesis (HDDS). The purpose of this study is to investigate the sagittal alignment of the cervical spine in HDDS and how the cervical spine responds to reduction of spondylolisthesis. Methods: Thirty-three adolescent patients with lumbar developmental spondylolisthesis who received preoperative and postoperative whole-spine x-rays were reviewed. They were divided into the HDDS group (n = 24, 13.0 ± 2.2 years old) and the low dysplastic developmental spondylolisthesis (LDDS) group (n = 9, 15.6 ± 1.9 years old). Spinal and pelvic sagittal parameters, including cervical lordosis (CL), were measured and compared between groups. In the HDDS group, the postoperative parameters were measured and compared with those before surgery. Results: HDDS group had a higher proportion of cervical kyphosis (70.8% vs. 22.2%, P = 0.019), and there was a significant difference in CL between the two groups (− 8.5° ± 16.1° vs. 10.5° ± 11.8°, P = 0.003). CL was correlated with the Dubousset’s lumbosacral angle (Dub-LSA), pelvic tilt (PT), and thoracic kyphosis (TK). -

Promising: Process Improvement in Psychosocial Health
PROMISing: Process Improvement in Psychosocial Health Carly Woodmark MS │ Dereesa Reid MBA │ Daniel Bouton MD SHC-Portland │ Department of Performance Improvement Abstract no. 20 Shriners Team And Patients PROMISing Changes Shriners Hospitals for Children is a network of 22 non-profit medical facilities across North America. Benefits of PROMIS Intervention Pre-op Post-op Since 1924, SHC-Portland has treated a wide range of pediatric orthopedic conditions, from fractures to rare diseases and syndromes. Our Integrated Practice Unit of multi-disciplinary Minor burden of taking PROMIS is offset by quality professionals provide a comprehensive approach through specialized evaluation and treatment communication of meaningful progress between along with rehabilitative services to restore each child physically, emotionally, and socially. Below is patient/family & physician during clinic visit. a list of common conditions treated at SHC-Portland. Medical providers can demonstrate improvements Skeletal abnormalities – Osteogenesis imperfecta (OI), osteochondritis dissecans (OCD lesions), from interventions & adjust care management if Blount disease, skeletal dysplasias, etc. needed. Outcome Performance Improvement Neuromuscular conditions – Cerebral palsy, myelomeningocele (spina bifida), Muscular dystrophy, spinal muscular atrophy After one year of data collection, rates of Minimal Clinical Important Difference (MCID) were assessed for all patient-reported domains in both surgical and non-surgical populations. Multivariate Hand/Upper extremity -

Sotos Syndrome
European Journal of Human Genetics (2007) 15, 264–271 & 2007 Nature Publishing Group All rights reserved 1018-4813/07 $30.00 www.nature.com/ejhg PRACTICAL GENETICS In association with Sotos syndrome Sotos syndrome is an autosomal dominant condition characterised by a distinctive facial appearance, learning disability and overgrowth resulting in tall stature and macrocephaly. In 2002, Sotos syndrome was shown to be caused by mutations and deletions of NSD1, which encodes a histone methyltransferase implicated in chromatin regulation. More recently, the NSD1 mutational spectrum has been defined, the phenotype of Sotos syndrome clarified and diagnostic and management guidelines developed. Introduction In brief Sotos syndrome was first described in 1964 by Juan Sotos Sotos syndrome is characterised by a distinctive facial and the major diagnostic criteria of a distinctive facial appearance, learning disability and childhood over- appearance, childhood overgrowth and learning disability growth. were established in 1994 by Cole and Hughes.1,2 In 2002, Sotos syndrome is associated with cardiac anomalies, cloning of the breakpoints of a de novo t(5;8)(q35;q24.1) renal anomalies, seizures and/or scoliosis in B25% of translocation in a child with Sotos syndrome led to the cases and a broad variety of additional features occur discovery that Sotos syndrome is caused by haploinsuffi- less frequently. ciency of the Nuclear receptor Set Domain containing NSD1 abnormalities, such as truncating mutations, protein 1 gene, NSD1.3 Subsequently, extensive analyses of missense mutations in functional domains, partial overgrowth cases have shown that intragenic NSD1 muta- gene deletions and 5q35 microdeletions encompass- tions and 5q35 microdeletions encompassing NSD1 cause ing NSD1, are identifiable in the majority (490%) of 490% of Sotos syndrome cases.4–10 In addition, NSD1 Sotos syndrome cases. -

Accelerating Matchmaking of Novel Dysmorphology Syndromes Through Clinical and Genomic Characterization of a Large Cohort
ORIGINAL RESEARCH ARTICLE © American College of Medical Genetics and Genomics Accelerating matchmaking of novel dysmorphology syndromes through clinical and genomic characterization of a large cohort Ranad Shaheen, PhD1, Nisha Patel, PhD1, Hanan Shamseldin, MSc1, Fatema Alzahrani, BSc1, Ruah Al-Yamany, MD1, Agaadir ALMoisheer, MSc1, Nour Ewida, BSc1, Shamsa Anazi, MSc1, Maha Alnemer, MD2, Mohamed Elsheikh, MD3, Khaled Alfaleh, MD3,4, Muneera Alshammari, MD4, Amal Alhashem, MD5, Abdullah A. Alangari, MD4, Mustafa A. Salih, MD4, Martin Kircher, MD6, Riza M. Daza, PhD6, Niema Ibrahim, BSc1, Salma M. Wakil, PhD1, Ahmed Alaqeel, MD7, Ikhlas Altowaijri, MD7, Jay Shendure, MD, PhD6, Amro Al-Habib, MD7, Eissa Faqieh, MD8 and Fowzan S. Alkuraya, MD1,9 Purpose: Dysmorphology syndromes are among the most common and C3ORF17). A significant minority of the phenotypes (6/31, 19%), referrals to clinical genetics specialists. Inability to match the dysmor- however, were caused by genes known to cause Mendelian pheno- phology pattern to a known syndrome can pose a major diagnostic chal- types, thus expanding the phenotypic spectrum of the diseases linked lenge. With an aim to accelerate the establishment of new syndromes to these genes. The conspicuous inheritance pattern and the highly and their genetic etiology, we describe our experience with multiplex specific phenotypes appear to have contributed to the high yield consanguineous families that appeared to represent novel autosomal (90%) of plausible molecular diagnoses in our study cohort. recessive dysmorphology syndromes at the time of evaluation. Conclusion: Reporting detailed clinical and genomic analysis of Methods: Combined autozygome/exome analysis of multiplex a large series of apparently novel dysmorphology syndromes will consanguineous families with apparently novel dysmorphology syn- likely lead to a trend to accelerate the establishment of novel syn- dromes.