Characterizing the Molecular Biology of a Bacteriophage-Like Particle from Bartonella Bacilliformis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Bartonella Apis Sp. Nov., a Honey Bee Gut Symbiont of the Class Alphaproteobacteria
Serveur Academique´ Lausannois SERVAL serval.unil.ch Author Manuscript Faculty of Biology and Medicine Publication This paper has been peer-reviewed but does not include the final publisher proof-corrections or journal pagination. Published in final edited form as: Title: Bartonella apis sp. nov., a honey bee gut symbiont of the class Alphaproteobacteria. Authors: Keˇsnerov´aL, Moritz R, Engel P Journal: International journal of systematic and evolutionary microbiology Year: 2016 Jan Issue: 66 Volume: 1 Pages: 414-21 DOI: 10.1099/ijsem.0.000736 In the absence of a copyright statement, users should assume that standard copyright protection applies, unless the article contains an explicit statement to the contrary. In case of doubt, contact the journal publisher to verify the copyright status of an article. 1 Bartonella apis sp. nov., a honey bee gut symbiont of the 2 class Alphaproteobacteria 3 4 Lucie Kešnerová, Roxane Moritz, Philipp Engel* 5 6 Department of Fundamental Microbiology, University of Lausanne, CH-1015 7 Lausanne, Switzerland 8 9 Running title: Description of a bee gut symbiont 10 11 *Correspondence: 12 Prof. Philipp Engel 13 Department of Fundamental Microbiology 14 University of Lausanne, CH-1015 Lausanne, Switzerland 15 Tel.: +41 (0)21 692 56 12 16 e-mail: [email protected] 17 18 Category: New Taxa – Proteobacteria 19 Keywords: Apis mellifera; insect; Bartonella; gut microbiota; Alpha-1 20 21 Sequence deposition: The 16S rRNA gene sequences and protein-coding gene 22 sequences of the bacterial strains PEB0122T, PEB0149, PEB0150, BBC0104, and 23 BBC0108 from Apis mellifera, and the uncultured Rhizobiales bacterium from 24 Herpagnathos saltator are deposited in GenBank with accession numbers KP987849 25 – KP987886 and KT315729 – KT315734. -

View Tickborne Diseases Sample Report
1360 Bayport Ave, Ste B. San Carlos, CA 94070 1(866) 364-0963 | [email protected] | www. vibrant-wellness.com PATIENT PROVIDER NAME: DEMO REPORT GENDER: Male PRACTICE NAME: Vibrant IT4 Practice DATE OF BIRTH: 04/14/1998 AGE: 22 PROVIDER NAME: Demo Client, DDD (999994) ADDRESS: TEST STREET, TEST CITY, KY- 42437. ACCESSION ID: 2009220006 PHLEBOTOMIST: 607 SPECIMEN COLLECTION TIME: 09-21-2020 11:14 SPECIMEN RECEIVED TIME: 09-22-2020 05:14 FINAL REPORT TIME: 09-25-2020 15:56 FASTING: FASTING Your Vibrant Wellness TickBorne 2.0 panel results are enclosed. These results are intended to aid in the diagnosis of tickborne diseases by your healthcare provider. The Vibrant Tickborne Diseases panel tests for IgG and IgM antibodies for Borreliosis/Lyme disease as well as co-infection(s) and opportunistic infections with other tick-borne illnesses along with detection of DNA of the species causing these infections. The Vibrant Immunochip test is a semiquantitative assay that detects IgG and IgM antibodies in human serum. The PCR Test is a real-time PCR Assay designed for qualitative detection of infectious group- specific DNA in clinical samples. Interpretation of Report: The test results of antibody levels to the individual antigens are calculated by comparing the average intensity of the individual antibody to that of a reference population and cut-off chosen for each protein. Reference ranges have been established using a well characterized set of more than 300 serum samples and antibodies to specific bacteria tested. The results are displayed as In Control, Moderate, or High Risk.for each antigen tested. -

Abstract Pultorak, Elizabeth Lauren
ABSTRACT PULTORAK, ELIZABETH LAUREN. The Epidemiology of Lyme Disease and Bartonellosis in Humans and Animals. (Under the direction of Edward B. Breitschwerdt). The expansion of vector borne diseases in humans, a variety of mammalian hosts, and arthropod vectors draws attention to the need for enhanced diagnostic techniques for documenting infection in hosts, effective vector control, and treatment of individuals with associated diseases. Through improved diagnosis of vector-borne disease in both humans and animals, epidemiological studies to elucidate clinical associations or spatio-temporal relationships can be assessed. Veterinarians, through the use of the C6 peptide in the SNAP DX test kit, may be able to evaluate the changing epidemiology of borreliosis through their canine population. We developed a survey to evaluate the practices and perceptions of veterinarians in North Carolina regarding borreliosis in dogs across different geographic regions of the state. We found that veterinarians’ perception of the risk of borreliosis in North Carolina was consistent with recent scientific reports pertaining to geographic expansion of borreliosis in the state. Veterinarians should promote routine screening of dogs for Borrelia burgdorferi exposure as a simple, inexpensive form of surveillance in this transitional geographic region. We next conducted two separate studies to evaluate Bartonella spp. bacteremia or presence of antibodies against B. henselae, B. koehlerae, or B. vinsonii subsp. berkhoffii in 296 patients examined by a rheumatologist and 192 patients with animal exposure (100%) and recent animal bites and scratches (88.0%). Among 296 patients examined by a rheumatologist, prevalence of antibodies (185 [62%]) and Bartonella spp. bacteremia (122 [41.1%]) was high. -

Detection and Partial Molecular Characterization of Rickettsia and Bartonella from Southern African Bat Species
Detection and partial molecular characterization of Rickettsia and Bartonella from southern African bat species by Tjale Mabotse Augustine (29685690) Submitted in partial fulfillment of the requirements for the degree MAGISTER SCIENTIAE (MICROBIOLOGY) in the Department of Microbiology and Plant Pathology Faculty of Natural and Agricultural Sciences University of Pretoria Pretoria, South Africa Supervisor: Dr Wanda Markotter Co-supervisors: Prof Louis H. Nel Dr Jacqueline Weyer May, 2012 I declare that the thesis, which I hereby submit for the degree MSc (Microbiology) at the University of Pretoria, South Africa, is my own work and has not been submitted by me for a degree at another university ________________________________ Tjale Mabotse Augustine i Acknowledgements I would like send my sincere gratitude to the following people: Dr Wanda Markotter (University of Pretoria), Dr Jacqueline Weyer (National Institute for Communicable Diseases-National Health Laboratory Service) and Prof Louis H Nel (University of Pretoria) for their supervision and guidance during the project. Dr Jacqueline Weyer (Centre for Zoonotic and Emerging diseases (Previously Special Pathogens Unit), National Institute for Communicable Diseases (National Heath Laboratory Service), for providing the positive control DNA for Rickettsia and Dr Jenny Rossouw (Special Bacterial Pathogens Reference Unit, National Institute for Communicable Diseases-National Health Laboratory Service), for providing the positive control DNA for Bartonella. Dr Teresa Kearney (Ditsong Museum of Natural Science), Gauteng and Northern Region Bat Interest Group, Kwa-Zulu Natal Bat Interest Group, Prof Ara Monadjem (University of Swaziland), Werner Marias (University of Johannesburg), Dr Francois du Rand (University of Johannesburg) and Prof David Jacobs (University of Cape Town) for collection of blood samples. -

Tick-Borne Pathogens in Removed Ticks Veneto, Northeastern Italy
Tick-borne pathogens in removed ticks Veneto, northeastern Italy: A cross-sectional investigation Anna Beltrame, Maureen Laroche, Monica Degani, Francesca Perandin, Zeno Bisoffi, Didier Raoult, Philippe Parola To cite this version: Anna Beltrame, Maureen Laroche, Monica Degani, Francesca Perandin, Zeno Bisoffi, et al.. Tick- borne pathogens in removed ticks Veneto, northeastern Italy: A cross-sectional investigation. Travel Medicine and Infectious Disease, Elsevier, 2018, 26, pp.58-61. 10.1016/j.tmaid.2018.08.008. hal- 01970220 HAL Id: hal-01970220 https://hal.archives-ouvertes.fr/hal-01970220 Submitted on 10 Apr 2019 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Travel Medicine and Infectious Disease 26 (2018) 58–61 Contents lists available at ScienceDirect Travel Medicine and Infectious Disease journal homepage: www.elsevier.com/locate/tmaid Tick-borne pathogens in removed ticks Veneto, northeastern Italy: A cross- sectional investigation T ∗ Anna Beltramea, , Maureen Larocheb, Monica Degania, Francesca Perandina, Zeno Bisoffia, Didier Raoultc, Philippe Parolab a Centre for Tropical Diseases, IRCCS Sacro Cuore Don Calabria Hospital, Via Sempreboni 5, 37024, Negrar, Italy b Aix Marseille Univ, AP-HM, SSA, VITROME, IHU-Méditerranée Infection, 19-21 Bd Jean Moulin, 13005, Marseille, France c Aix Marseille Univ, AP-HM, MEPHI, IHU-Méditerranée Infection, 19-21 Bd Jean Moulin, 13005, Marseille, France ARTICLE INFO ABSTRACT Keywords: Background: In Italy, the incidence of tick-borne diseases in humans is underestimated, as they are not ob- Tick-borne diseases ligatorily notifiable. -

Co-Infection of Bacteria and Protozoan Parasites in Ixodes Ricinus Nymphs Collected in the Alsace Region, France
Ticks and Tick-borne Diseases 10 (2019) 101241 Contents lists available at ScienceDirect Ticks and Tick-borne Diseases journal homepage: www.elsevier.com/locate/ttbdis Short communication Co-infection of bacteria and protozoan parasites in Ixodes ricinus nymphs T collected in the Alsace region, France Amira Nebbaka,b, Handi Dahmanaa, Lionel Almerasa,c, Didier Raoultd, Nathalie Boulangere,f, ⁎ Benoit Jaulhace,f, Oleg Mediannikovd, Philippe Parolaa,d, a Aix Marseille Univ, IRD, AP-HM, SSA, VITROME, Marseille, France b Centre de Recherche Scientifique et Technique en Analyses Physico-Chimiques (CRAPC). Zone Industrielle, BP 384 Bou-Ismail, Tipaza Algeria c Unité de Parasitologie et Entomologie, Département des Maladies Infectieuses, Institut de Recherche Biomédicale des Armées, Marseille, France d IHU-Méditerranée Infection, Marseille, France e Centre National de Reference Borrelia, Centre Hospitalier Universitaire, Strasbourg, France f EA7290Virulence bactérienne précoce: groupe borréliose de Lyme, Facultés de pharmacie et de médecine, Université de Strasbourg, France ARTICLE INFO ABSTRACT Keywords: Fifty nymphal Ixodes ricinus ticks collected in Alsace, France, identified by morphological criteria and using Ixodes ricinus MALDI-TOF MS, were tested by PCR to detect tick-associated bacteria and protozoan parasites. Seventy percent Co-infection (35/50) of ticks contained at least one microorganism; 26% (9/35) contained two or more species. Several Bacterial pathogens human pathogens were identified including Borrelia burgdorferi s.s. (4%), Borrelia afzelii (2%), Borrelia garinii Protozoan parasites (2%), Borrelia valaisiana (4%), Borrelia miyamotoi (2%), Rickettsia helvetica (6%) and “Babesia venatorum” (2%). MALDI-TOF MS Bartonella spp. (10%) and a Wolbachia spp. (8%) were also detected. The most common co-infections involved Anaplasmataceae with Borrelia spp. -

Bartonella: Feline Diseases and Emerging Zoonosis
BARTONELLA: FELINE DISEASES AND EMERGING ZOONOSIS WILLIAM D. HARDY, JR., V.M.D. Director National Veterinary Laboratory, Inc. P.O Box 239 Franklin Lakes, New Jersey 07417 201-891-2992 www.natvetlab.com or .net Gingivitis Proliferative Gingivitis Conjunctivitis/Blepharitis Uveitis & Conjunctivitis URI Oral Ulcers Stomatitis Lymphadenopathy TABLE OF CONTENTS Page SUMMARY……………………………………………………………………………………... i INTRODUCTION……………………………………………………………………………… 1 MICROBIOLOGY……………………………………………………………………………... 1 METHODS OF DETECTION OF BARTONELLA INFECTION.………………………….. 1 Isolation from Blood…………………………………………………………………….. 2 Serologic Tests…………………………………………………………………………… 2 SEROLOGY……………………………………………………………………………………… 3 CATS: PREVALENCE OF BARTONELLA INFECTIONS…………………………………… 4 Geographic Risk factors for Infection……………………………………………………. 5 Risk Factors for Infection………………………………………………………………… 5 FELINE BARTONELLA DISEASES………………………………………………………….… 6 Bartonella Pathogenesis………………………………………………………………… 7 Therapy of Feline Bartonella Diseases…………………………………………………… 14 Clinical Therapy Results…………………………………………………………………. 15 DOGS: PREVALENCE OF BARTONELLA INFECTIONS…………………………………. 17 CANINE BARTONELLA DISEASES…………………………………………………………... 17 HUMAN BARTONELLA DISEASES…………………………………………………………… 18 Zoonotic Case Study……………………………………………………………………... 21 FELINE BLOOD DONORS……………………………………………………………………. 21 REFERENCES………………………………………………………………………………….. 22 This work was initiated while Dr. Hardy was: Professor of Medicine, Albert Einstein College of Medicine of Yeshiva University, Bronx, New York and Director, -
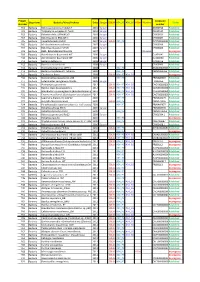
Project Number Organisms Bacteria/Virus/Archaea Date
Project_ Accession Organisms Bacteria/Virus/Archaea Date Sanger SOLiD 454_PE 454_SG PGM Illumina Status Number number P01 Bacteria Rickettsia conorii str.Malish 7 2001 Sanger AE006914 Published P02 Bacteria Tropheryma whipplei str.Twist 2003 Sanger AE014184 Published P03 Bacteria Rickettsia felis URRWXCal2 2005 Sanger CP000053 Published P04 Bacteria Rickettsia bellii RML369-C 2006 Sanger CP000087 Published P05 Bacteria Coxiella burnetii CB109 2007 Sanger SOLiD 454_PE AKYP00000000 Published P06 Bacteria Minibacterium massiliensis 2007 Sanger CP000269 Published P07 Bacteria Rickettsia massiliae MTU5 2007 Sanger CP000683 Published P08 Bacteria BaBL=Bête à Bernard Lascola 2007 Illumina In progress P09 Bacteria Acinetobacter baumannii AYE 2006 Sanger CU459141 Published P10 Bacteria Acinetobacter baumannii SDF 2006 Sanger CU468230 Published P11 Bacteria Borrelia duttonii Ly 2008 Sanger CP000976 Published P12 Bacteria Borrelia recurrentis A1 2008 Sanger CP000993 Published P13 Bacteria Francisella tularensis URFT1 2008 454_PE ABAZ00000000Published P14 Bacteria Borrelia crocidurae str. Achema 2009 454_PE PRJNA162335 Published P15 Bacteria Citrobacter koseri 2009 SOLiD 454_PE 454_SG In progress P16 Bacteria Diplorickettsia massiliensis 20B 2009 454_PE PRJNA86907 Published P17 Bacteria Enterobacter aerogenes EA1509E 2009 Sanger FO203355 Published P18 Bacteria Actinomyces grossensis 2012 SOLiD 454_PE 454_SG CAGY00000000Published P19 Bacteria Bacillus massiliosenegalensis 2012 SOLiD 454_PE 454_SG CAHJ00000000 Published P20 Bacteria Brevibacterium senegalensis -

Human Bartonellosis: an Underappreciated Public Health Problem?
Tropical Medicine and Infectious Disease Review Human Bartonellosis: An Underappreciated Public Health Problem? Mercedes A. Cheslock and Monica E. Embers * Division of Immunology, Tulane National Primate Research Center, Tulane University Health Sciences, Covington, LA 70433, USA; [email protected] * Correspondence: [email protected]; Tel.: +(985)-871-6607 Received: 24 March 2019; Accepted: 16 April 2019; Published: 19 April 2019 Abstract: Bartonella spp. bacteria can be found around the globe and are the causative agents of multiple human diseases. The most well-known infection is called cat-scratch disease, which causes mild lymphadenopathy and fever. As our knowledge of these bacteria grows, new presentations of the disease have been recognized, with serious manifestations. Not only has more severe disease been associated with these bacteria but also Bartonella species have been discovered in a wide range of mammals, and the pathogens’ DNA can be found in multiple vectors. This review will focus on some common mammalian reservoirs as well as the suspected vectors in relation to the disease transmission and prevalence. Understanding the complex interactions between these bacteria, their vectors, and their reservoirs, as well as the breadth of infection by Bartonella around the world will help to assess the impact of Bartonellosis on public health. Keywords: Bartonella; vector; bartonellosis; ticks; fleas; domestic animals; human 1. Introduction Several Bartonella spp. have been linked to emerging and reemerging human diseases (Table1)[ 1–5]. These fastidious, gram-negative bacteria cause the clinically complex disease known as Bartonellosis. Historically, the most common causative agents for human disease have been Bartonella bacilliformis, Bartonella quintana, and Bartonella henselae. -

Bartonella Talpae Comb
INTERNATIONAL JOURNALOF SYSTEMATIC BACTERIOLOGY, Jan. 1995, p. 1-8 Vol. 45, No. 1 0020-7713/95/$04.00+0 Copyright 0 1995, International Union of Microbiological Societies Proposals To Unify the Genera Grahamella and Bartonella, with Descriptions of Bartonella talpae comb. nov., Bartonella peromysci comb. nov., and Three New Species, Bartonella grahamii sp. nov., Bartonella taylorii sp. nov., and Bartonella doshiae sp. nov. RICHARD J. BIRTLES,’ * TIMOTHY G. HARRISON,2 NICHOLAS A. SAUNDERS,2 AND DAVID H. MOLYNEUX3 Respiratory and Systemic Infection Laboratory, and Laboratory of Microbiological Reagents, Central Public Health Laboratory, London Nw9 5HT and School of Tropical Medicine, Liverpool L3 SQA, United Kingdom Polyphasic methods were used to examine the taxonomic positions of three newly identified Grahamella species. A comparison of the 16s rRNA gene sequences of these organisms with the sequences available for other bacteria revealed that these three species form a tight monophyletic cluster with members of the genus Bartonella. This cluster is only remotely related to other members of the order Rickettsiales. Determinations of the levels of DNA relatedness between Grahamella species and Bartonella species (by using a modified hydroxyapatite method) revealed that all of the species belonging to these two genera are distinct but closely related. On the basis of these data and the results of guanine-plus-cytosine content and phenotypic characterization studies, we propose that the genera Grahumella and Bartonella should be unified and that the latter name should be retained. Bartonella talpae and Bartonella peromysci, new combinations for former Grahamella species, are created, and the following three new Bartonella species are described: Bartonella grahamii, Bartonella taylorii, and Bartonella doshiae. -

BARTONELLOSIS: a ONE HEALTH PERSPECTIVES on an EMERGING INFECTIOUS DISEASE Edward B
BARTONELLOSIS: A ONE HEALTH PERSPECTIVES ON AN EMERGING INFECTIOUS DISEASE Edward B. Breitschwerdt, DVM, DACVIM (Small Animal Internal Medicine) College of Veterinary Medicine, North Carolina State University, Raleigh, NC INTRODUCTION Bartonella species are fastidious Gram-negative bacteria that are highly adapted to a mammalian reservoir host and within which the bacteria usually cause a long-lasting intraerythrocytic bacteremia.1-3 These facts are of particular importance to veterinarians and physicians, as an increasing number of animal reservoir hosts have been identified for various Bartonella species. Among numerous other examples, Bartonella henselae has co-evolved with cats, Bartonella vinsonii subsp. berkhoffii has co-evolved with dogs and wild canines, and Bartonella bovis has co-evolved with cattle. Importantly, the list of reservoir-adapted Bartonella species, including a large number of rodent species that might serve as “pocket pets”, continues to grow exponentially, as new Bartonella spp. are discovered.2-3 Prior to 1990, there were only two named Bartonella species, whereas there are now at least 35 named and numerous unnamed or candidatus species, based upon deposited GenBank sequences or preliminary reports, respectively. In the natural reservoir host, chronic bacteremia with a Bartonella species can frequently be detected by blood culture or PCR in outwardly healthy individuals. In contrast, the diagnostic detection of a Bartonella spp. in a non-reservoir adapted host can be extremely difficult. Most, although not all diseases caused by Bartonella spp., occur in accidental hosts and these organisms are being increasingly implicated as a cause of zoonotic infections.4-8 Until recently, mechanisms that facilitate persistent Bartonella bacteremia in mammals were not well understood. -

Bartonella Gabonensis Sp. Nov., a New Bartonella Species from Savannah Rodent Lophuromys Sp
TAXONOGENOMICS: GENOME OF A NEW ORGANISM Bartonella gabonensis sp. nov., a new bartonella species from savannah rodent Lophuromys sp. in Franceville, Gabon J. B. Mangombi1,3,4,N.N’Dilimabaka1,2, H. Medkour4,5, O. L. Banga1, M. L. Tall4,5, M. Ben Khedher4,5, J. Terras4,5, S. Abdi4,5, M. Bourgarel6,7, E. Leroy8, F. Fenollar3,4 and O. Mediannikov4,5 1) Centre Interdisciplinaire de Recherches Médicales de Franceville (CIRMF), 2) Département de Biologie, Université des Sciences et Techniques de Masuku (USTM), Franceville, Gabon, 3) Aix-Marseille Université, IRD, APHM, Microbes, VITROME, 4) IHU Méditerranée Infection, 5) Aix-Marseille Université, IRD, APHM, Microbes, MEPHI, Marseille, France, 6) ASTRE, Université Montpellier, CIRAD, INRA, 7) UMR MIVEGEC IRDCNRSUM, Institut de Recherche pour le Développement (IRD), Montpellier, France and 8) CIRAD, UMR ASTRE, Harare, Zimbabwe Abstract We describe a new strain named Bartonella gabonensis sp. nov. strain 669T (CSURB1083). The entire genome of this strain is described here. It was isolated from a savannah rodent, a brush-furred rat (Lophuromys sp.), trapped the city of Franceville in Gabon, in Central Africa. B. gabonensis is an aerobic, rod-shaped and Gram-negative bacterium. On the basis of the organism’s features, and following a taxonogenomic approach, we propose the creation of the species Bartonella gabonensis sp. nov. © 2020 The Authors. Published by Elsevier Ltd. Keywords: Bartonella gabonensis sp. nov., Gabon, genome, Lophuromys sp., rodents Original Submission: 20 June 2020; Revised Submission: 7 October 2020; Accepted: 14 October 2020 Article published online: 27 October 2020 The latest epidemiologic studies from around the world have Corresponding author: N.