Roles for the Aryl Hydrocarbon Receptor in the Immune Response to Toxoplasma Gondii
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Epigenetic Regulations of Ahr in the Aspect of Immunomodulation
International Journal of Molecular Sciences Review Epigenetic Regulations of AhR in the Aspect of Immunomodulation Anna Wajda 1,* , Joanna Łapczuk-Roma ´nska 2 and Agnieszka Paradowska-Gorycka 1 1 Department of Molecular Biology, National Institute of Geriatrics, Rheumatology and Rehabilitation, 02-637 Warsaw, Poland; [email protected] 2 Department of Experimental and Clinical Pharmacology, Pomeranian Medical University, 70-111 Szczecin, Poland; [email protected] * Correspondence: [email protected] Received: 31 July 2020; Accepted: 28 August 2020; Published: 3 September 2020 Abstract: Environmental factors contribute to autoimmune disease manifestation, and as regarded today, AhR has become an important factor in studies of immunomodulation. Besides immunological aspects, AhR also plays a role in pharmacological, toxicological and many other physiological processes such as adaptive metabolism. In recent years, epigenetic mechanisms have provided new insight into gene regulation and reveal a new contribution to autoimmune disease pathogenesis. DNA methylation, histone modifications, chromatin alterations, microRNA and consequently non-genetic changes in phenotypes connect with environmental factors. Increasing data reveals AhR cross-roads with the most significant in immunology pathways. Although study on epigenetic modulations in autoimmune diseases is still not well understood, therefore future research will help us understand their pathophysiology and help to find new therapeutic strategies. Present literature review -

Environmental Pollution 275 (2021) 116665
Environmental Pollution 275 (2021) 116665 Contents lists available at ScienceDirect Environmental Pollution journal homepage: www.elsevier.com/locate/envpol Metabolomics reveals the reproductive abnormality in female zebrafish exposed to environmentally relevant levels of climbazole* Ting Zou a, 1, Yan-Qiu Liang b, 1, Xiaoliang Liao a, Xiao-Fan Chen a, Tao Wang c, * Yuanyuan Song c, Zhi-Cheng Lin a, Zenghua Qi a, Zhi-Feng Chen a, d, , Zongwei Cai a, c a Guangdong Key Laboratory of Environmental Catalysis and Health Risk Control, School of Environmental Science and Engineering, Institute of Environmental Health and Pollution Control, Guangdong University of Technology, Guangzhou, 510006, China b Faculty of Chemistry and Environmental Science, Guangdong Ocean University, Zhanjiang, 524088, China c State Key Laboratory of Environmental and Biological Analysis, Department of Chemistry, Hong Kong Baptist University, Hong Kong Special Administrative Region, China d Guangdong Provincial Key Laboratory of Chemical Pollution and Environmental Safety, South China Normal University, Guangzhou, 510006, China article info abstract Article history: Climbazole (CBZ) ubiquitously detected in the aquatic environment may disrupt fish reproductive Received 15 November 2020 function. Thus far, the previous study has focused on its transcriptional impact of steroidogenesis-related Received in revised form genes on zebrafish, but the underlying toxic mechanism still needs further investigation at the metabolic 10 January 2021 level. In this study, adult zebrafish were chronically exposed to CBZ at concentrations of 0.1 (corre- Accepted 2 February 2021 sponding to the real concentration in surface water), 10, and 1000 mg/L and evaluated for reproductive Available online 5 February 2021 function by egg production, with subsequent ovarian tissue samples taken for histology, metabolomics, and other biochemical analysis. -

Role of Arachidonic Acid and Its Metabolites in the Biological and Clinical Manifestations of Idiopathic Nephrotic Syndrome
International Journal of Molecular Sciences Review Role of Arachidonic Acid and Its Metabolites in the Biological and Clinical Manifestations of Idiopathic Nephrotic Syndrome Stefano Turolo 1,* , Alberto Edefonti 1 , Alessandra Mazzocchi 2, Marie Louise Syren 2, William Morello 1, Carlo Agostoni 2,3 and Giovanni Montini 1,2 1 Fondazione IRCCS Ca’ Granda-Ospedale Maggiore Policlinico, Pediatric Nephrology, Dialysis and Transplant Unit, Via della Commenda 9, 20122 Milan, Italy; [email protected] (A.E.); [email protected] (W.M.); [email protected] (G.M.) 2 Department of Clinical Sciences and Community Health, University of Milan, 20122 Milan, Italy; [email protected] (A.M.); [email protected] (M.L.S.); [email protected] (C.A.) 3 Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, Pediatric Intermediate Care Unit, 20122 Milan, Italy * Correspondence: [email protected] Abstract: Studies concerning the role of arachidonic acid (AA) and its metabolites in kidney disease are scarce, and this applies in particular to idiopathic nephrotic syndrome (INS). INS is one of the most frequent glomerular diseases in childhood; it is characterized by T-lymphocyte dysfunction, alterations of pro- and anti-coagulant factor levels, and increased platelet count and aggregation, leading to thrombophilia. AA and its metabolites are involved in several biological processes. Herein, Citation: Turolo, S.; Edefonti, A.; we describe the main fields where they may play a significant role, particularly as it pertains to their Mazzocchi, A.; Syren, M.L.; effects on the kidney and the mechanisms underlying INS. AA and its metabolites influence cell Morello, W.; Agostoni, C.; Montini, G. -

Lipoxin A4: a Novel Anti-Inflammatory Molecule? Thorax: First Published As 10.1136/Thx.50.2.111 on 1 February 1995
Thorax 1995;50:111-112 illl Lipoxin A4: a novel anti-inflammatory molecule? Thorax: first published as 10.1136/thx.50.2.111 on 1 February 1995. Downloaded from Arachidonic acid is metabolised by the cyclooxygenase contractions.22 These studies support the view that LXA4 pathway to the prostaglandins and thromboxane A2 or via may act as a partial agonist at the same or similar site as one of the lipoxygenase pathways.' Three major lip- the sulphidopeptide leukotrienes. oxygenase pathways have been identified in mammalian The fact that 1 5-lipoxygenase is abundant in lung tissue tissue - namely, the 5-, 12-, and 15-lipoxygenases.2A The and that LXA4 has been recovered in the bronchoalveolar 5-lipoxygenase pathway metabolises arachidonic acid lavage fluid ofpatients with asthma and other lung diseases through two intermediates, 5-hydroperoxyeicosatetranoic suggests that LXA4 may be a potential mediator or mod- acid (5-HPETE) and leukotriene A4 (LTA4), to LTB4 and ulator of inflammation in the lung. In a recent study eight the sulphidopeptide leukotrienes LTC4, LTD4, and LTE4.5 subjects underwent inhalation challenge with LXA4,23 but The sulphidopeptide leukotrienes are potent spasmogens6 no effect was seen on specific conductance, rate of airflow for non-vascular smooth muscle and may play a part in at 25% vital capacity (V25), blood pressure, pulse, or the pathogenesis of bronchial asthma.7`10 asthmatic symptoms. There was, however, a significant The interactions between 5-lipoxygenase and 15-lip- shift of the specific conductance and V25 dose-response oxygenase on arachidonic acid metabolism have recently curve to the right after inhalation challenge with LTC4 been studied and a new series of biologically active me- combined with LXA4 compared with that after inhalation tabolites described." 12 These molecules have been termed challenge with LTC4 alone. -

Reduced 15-Lipoxygenase 2 and Lipoxin A4/Leukotriene B4 Ratio in Children with Cystic Fibrosis
ORIGINAL ARTICLE CYSTIC FIBROSIS Reduced 15-lipoxygenase 2 and lipoxin A4/leukotriene B4 ratio in children with cystic fibrosis Fiona C. Ringholz1, Paul J. Buchanan1, Donna T. Clarke1, Roisin G. Millar1, Michael McDermott2, Barry Linnane1,3,4, Brian J. Harvey5, Paul McNally1,2 and Valerie Urbach1,6 Affiliations: 1National Children’s Research Centre, Crumlin, Dublin, Ireland. 2Our Lady’s Children’s Hospital, Crumlin, Dublin, Ireland. 3Midwestern Regional Hospital, Limerick, Ireland. 4Centre for Interventions in Infection, Inflammation and Immunity (4i), Graduate Entry Medical School, University of Limerick, Limerick, Ireland. 5Molecular Medicine Laboratories, Royal College of Surgeons in Ireland, Beaumont Hospital, Dublin, Ireland. 6Institut National de la Sante´ et de la Recherche Me´dicale, U845, Faculte´ de Me´decine Paris Descartes, Paris, France. Correspondence: Valerie Urbach, National Children’s Research Centre, Crumlin, Dublin 12, Ireland. E-mail: [email protected] ABSTRACT Airway disease in cystic fibrosis (CF) is characterised by impaired mucociliary clearance, persistent bacterial infection and neutrophilic inflammation. Lipoxin A4 (LXA4) initiates the active resolution of inflammation and promotes airway surface hydration in CF models. 15-Lipoxygenase (LO) plays a central role in the ‘‘class switch’’ of eicosanoid mediator biosynthesis from leukotrienes to lipoxins, initiating the active resolution of inflammation. We hypothesised that defective eicosanoid mediator class switching contributes to the failure to resolve inflammation in CF lung disease. Using bronchoalveolar lavage (BAL) samples from 46 children with CF and 19 paediatric controls we demonstrate that the ratio of LXA4 to leukotriene B4 (LTB4) is depressed in CF BAL (p,0.01), even in the absence of infection (p,0.001). -

Strict Regio-Specificity of Human Epithelial 15-Lipoxygenase-2
Strict Regio-specificity of Human Epithelial 15-Lipoxygenase-2 Delineates its Transcellular Synthesis Potential Abigail R. Green, Shannon Barbour, Thomas Horn, Jose Carlos, Jevgenij A. Raskatov, Theodore R. Holman* Department Chemistry and Biochemistry, University of California Santa Cruz, 1156 High Street, Santa Cruz CA 95064, USA *Corresponding author: Tel: 831-459-5884. Email: [email protected] FUNDING: This work was supported by the NIH NS081180 and GM56062. Abbreviations: LOX, lipoxygenase; h15-LOX-2, human epithelial 15-lipoxygenase-2; h15-LOX-1, human reticulocyte 15-lipoxygenase-1; sLO-1, soybean lipoxygenase-1; 5-LOX, leukocyte 5-lipoxygenase; 12-LOX, human platelet 12-lipoxygenase; GP, glutathione peroxidase; AA, arachidonic acid; HETE, hydoxy-eicosatetraenoic acid; HPETE, hydroperoxy-eicosatetraenoic acid; diHETEs, dihydroxy-eicosatetraenoic acids; 5-HETE, 5-hydroxy-6E,8Z,11Z,14Z-eicosatetraenoic acid; 5-HPETE, 5-hydro peroxy-6E,8Z,11Z,14Z-eicosatetraenoic acid; 12-HPETE, 12-hydroperoxy-5Z,8Z,10E, 14Z-eicosatetraenoic acid; 15-HPETE, 15-hydroperoxy-5Z,8Z,10Z,13E- eicosatetraenoic acid; 5,15-HETE, 5S,15S-dihydroxy-6E,8Z,10Z,13E-eicosatetraenoic acid; 5,15-diHPETE, 5,15-dihydroperoxy-6E,8Z,10Z,13E-eicosatetraenoic acid; 5,6- diHETE, 5S,6R-dihydroxy-7E,9E,11Z,14Z-eicosatetraenoic acid; LTA4, 5S-trans-5,6- oxido-7E,9E,11Z,14Z-eicosatetraenoic acid; LTB4, 5S,12R-dihydroxy-6Z,8E,10E,14Z- eicosatetraenoic acid; LipoxinA4 (LxA4), 5S,6R,15S-trihydroxy-7E,9E,11Z,13E- eicosatetraenoic acid; LipoxinB4 (LxB4), 5S,14R,15S-trihydroxy-6E,8Z,10E,12E- eicosatetraenoic acid. Abstract Lipoxins are an important class of lipid mediators that induce the resolution of inflammation, and arise from transcellular exchange of arachidonic acid (AA)- derived lipoxygenase products. -

Phospholipase A2 Regulates Eicosanoid Class Switching During Inflammasome Activation
Phospholipase A2 regulates eicosanoid class switching during inflammasome activation Paul C. Norrisa, David Gosselinb, Donna Reichartb, Christopher K. Glassb, and Edward A. Dennisa,1 Departments of aChemistry/Biochemistry and Pharmacology, and bCellular and Molecular Medicine, University of California, San Diego, La Jolla, CA 92093 Edited by Michael A. Marletta, The Scripps Research Institute, La Jolla, CA, and approved July 30, 2014 (received for review March 13, 2014) Initiation and resolution of inflammation are considered to be to initiate pathogenic killing, subsequent “class switching” to lipoxin tightly connected processes. Lipoxins (LX) are proresolution lipid (LX) formation by “reprogrammed” neutrophils inhibits additional mediators that inhibit phlogistic neutrophil recruitment and pro- neutrophil recruitment during self-resolving inflammatory resolu- mote wound-healing macrophage recruitment in humans via tion (9). The direct link between inflammatory commitment and potent and specific signaling through the LXA4 receptor (ALX). resolution mediated by eicosanoid signaling in macrophages One model of lipoxin biosynthesis involves sequential metabolism remains unclear from short-term vs. long-term priming, but the of arachidonic acid by two cell types expressing a combined trans- complete temporal changes and important interconnections cellular metabolon. It is currently unclear how lipoxins are effi- within the entire eicosadome are now demonstrated. ciently formed from precursors or if they are directly generated after receptor-mediated inflammatory commitment. Here, we pro- Results vide evidence for a pathway by which lipoxins are generated in We first primed immortalized macrophage-like cells (RAW264.7) macrophages as a consequence of sequential activation of toll-like with the TLR4 agonist Kdo2 lipid A (KLA) for various times and receptor 4 (TLR4), a receptor for endotoxin, and P2X7, a purinergic examined the effects on subsequent purinergic stimulated COX receptor for extracellular ATP. -

Identification of Potential Aryl Hydrocarbon Receptor Ligands by Virtual Screening of Industrial Chemicals
Chemical Research in Toxicology This document is confidential and is proprietary to the American Chemical Society and its authors. Do not copy or disclose without written permission. If you have received this item in error, notify the sender and delete all copies. Identification of potential aryl hydrocarbon receptor ligands by virtual screening of industrial chemicals Journal: Chemical Research in Toxicology Manuscript ID tx-2016-004609 Manuscript Type: Article Date Submitted by the Author: 20-Dec-2016 Complete List of Authors: Larsson, Malin; Umeå University, Department of Chemistry Fraccalvieri, Domenico; Università degli Studi di Milano-Bicocca, 1Dipartimento di Scienze dell’Ambiente e del Territorio Andersson, C. David; Umeå University, Department of Chemistry Bonati, Laura; University of Milano-Bicocca, Department of Earth and Environmental Sciences Linusson, Anna; Umeå University, Department of Chemistry Andersson, Patrik; Umea University, Department of Chemistry ACS Paragon Plus Environment Page 1 of 52 Chemical Research in Toxicology 1 2 3 4 Identification of potential aryl hydrocarbon receptor ligands by virtual 5 6 screening of industrial chemicals 7 8 9 10 Malin Larsson†, Domenico Fraccalvieri‡, C. David Andersson†, Laura Bonati‡, Anna Linusson†, and 11 12 Patrik L. Andersson†* 13 14 15 †Department of Chemistry, Umeå University, SE-901 87 Umeå, Sweden 16 17 18 ‡Department of Earth and Environmental Sciences, University of Milano-Bicocca, Piazza della Scienza 1, 19 20 21 20126 Milano, Italy 22 23 24 *Corresponding author: Tel.: +46-90-786-5266; fax: +46-90-786-7655. Email address: [email protected] 25 26 27 28 29 30 Malin Larsson†: [email protected] 31 32 33 ‡ 34 Domenico Fraccalvieri : [email protected] 35 36 † 37 C. -

Therapeutic Effects of Specialized Pro-Resolving Lipids Mediators On
antioxidants Review Therapeutic Effects of Specialized Pro-Resolving Lipids Mediators on Cardiac Fibrosis via NRF2 Activation 1, 1,2, 2, Gyeoung Jin Kang y, Eun Ji Kim y and Chang Hoon Lee * 1 Lillehei Heart Institute, University of Minnesota, Minneapolis, MN 55455, USA; [email protected] (G.J.K.); [email protected] (E.J.K.) 2 College of Pharmacy, Dongguk University, Seoul 04620, Korea * Correspondence: [email protected]; Tel.: +82-31-961-5213 Equally contributed. y Received: 11 November 2020; Accepted: 9 December 2020; Published: 10 December 2020 Abstract: Heart disease is the number one mortality disease in the world. In particular, cardiac fibrosis is considered as a major factor causing myocardial infarction and heart failure. In particular, oxidative stress is a major cause of heart fibrosis. In order to control such oxidative stress, the importance of nuclear factor erythropoietin 2 related factor 2 (NRF2) has recently been highlighted. In this review, we will discuss the activation of NRF2 by docosahexanoic acid (DHA), eicosapentaenoic acid (EPA), and the specialized pro-resolving lipid mediators (SPMs) derived from polyunsaturated lipids, including DHA and EPA. Additionally, we will discuss their effects on cardiac fibrosis via NRF2 activation. Keywords: cardiac fibrosis; NRF2; lipoxins; resolvins; maresins; neuroprotectins 1. Introduction Cardiovascular disease is the leading cause of death worldwide [1]. Cardiac fibrosis is a major factor leading to the progression of myocardial infarction and heart failure [2]. Cardiac fibrosis is characterized by the net accumulation of extracellular matrix proteins in the cardiac stroma and ultimately impairs cardiac function [3]. Therefore, interest in substances with cardioprotective activity continues. -
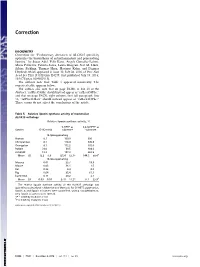
Evolutionary Alteration of ALOX15 Specificity Optimizes The
Correction BIOCHEMISTRY Correction for “Evolutionary alteration of ALOX15 specificity optimizes the biosynthesis of antiinflammatory and proresolving lipoxins,” by Susan Adel, Felix Karst, Àngels González-Lafont, Mária Pekárová, Patricia Saura, Laura Masgrau, José M. Lluch, Sabine Stehling, Thomas Horn, Hartmut Kuhn, and Dagmar Heydeck, which appeared in issue 30, July 26, 2016, of Proc Natl Acad Sci USA (113:E4266–E4275; first published July 13, 2016; 10.1073/pnas.1604029113). The authors note that Table 5 appeared incorrectly. The corrected table appears below. The authors also note that on page E4266, in line 20 of the Abstract, “ratPhe353Ala” should instead appear as “ratLeu353Phe;” and that on page E4270, right column, first full paragraph, line 12, “ratPhe353Leu” should instead appear as “ratLeu353Phe.” These errors do not affect the conclusions of the article. Table 5. Relative lipoxin synthase activity of mammalian ALOX15 orthologs Relative lipoxin synthase activity, % 5-HETE as 5,6-DiHETE as Species 15-/12-ratio substrate substrate 15-lipoxygenating Human 8.1 100.0 100 Chimpanzee 8.1 118.0 145.8 Orangutan 8.1 172.2 105.6 Rabbit 24.0 39.5 108.6 ratL353F 13.3 197.3 262.5 Mean ± SD 12.3 ± 6.9 125.4 ± 62.1* 144.5 ± 68.4† 12-lipoxygenating Macaca 0.01 25.7 19.9 Mouse 0.03 36.1 1.5 Rat 0.26 8.4 0.0 Pig 0.04 35.4 61.1 humI418A 0.11 29.2 2.1 Mean ± SD 0.09 ± 0.10 27.0 ± 11.2* 17.1 ± 25.9† The relative lipoxin synthase activity of the ALOX15 orthologs was quantified as described in Materials and Methods. -

New Series of Lipoxins Isolated from Human Eosinophils
Volume 255, number 1, 143-148 FEBS 07584 September 1989 New series of lipoxins isolated from human eosinophils Dieter Steinhilber and Hermann J. Roth Department of Pharmaceutical Chemistry, Pharmaceutical Institute, Auf der Morgenstelle 8, D-7400 Tiibingen, FRG Received 6 July 1989 Granulocytes from human eosinophilic donors were incubated with arachidonic acid or 15-hydroxyeicosatetraenoic acid (15-HETE) and stimulated with the ionophore A23187. The eicosanoids were extracted with reversed-phase cartridges and subjected to RP-HPLC analysis. When extracts from eosinophii-enriehed populations were analysed and compared with extracts from human neutrophils, three additional peaks were detected which coeluted with 15-hydroxy-z/13-trans- 15H derivatives of leukotriene C4, D 4 and E, in different HPLC systems. The recorded absorbance spectra of the eluted compounds and the standards were identical and showed a maximum at 307 nm which is characteristic for a conjugated tetraene system with a bathochromic shift by the sulfur moiety in ~t-position to the tetraene system. The compound which coeluted with the 15-hydroxy-LTC4 standard was treated with y-glutamyltransferase and converted to the corresponding leukotriene D 4 derivative. The results indicate that interaction between the 5- and 15-1ipoxygenase pathways leads to the formation of a new series of arachidonie acid metabolites in human eosinophils. Since the biosynthetic route is similar to that of lipoxin A, and lipoxin B4, we suggest the trivial names lipoxin C4, D 4 and E~. Lipoxin; Leukotriene; Lipoxygenase; Eosinophil; Inflammation 1. INTRODUCTION (15S)-hydroxy-5,6-epoxy-7,9,13-trans-11-cis- eicosatetraenoic acid was suggested to be the com- Recently, a new series of arachidonic acid me- mon intermediate in the biosynthesis of lipoxin A4 tabolites was reported which is produced by the in- and B4 in human granulocytes [3,4] and LXA4 was teraction of the 5- and the 15-1ipoxygenase path- found in extracts from human eosinophils way. -

Inflammation, Cancer and Oxidative Lipoxygenase Activity Are Intimately Linked
Cancers 2014, 6, 1500-1521; doi:10.3390/cancers6031500 OPEN ACCESS cancers ISSN 2072-6694 www.mdpi.com/journal/cancers Review Inflammation, Cancer and Oxidative Lipoxygenase Activity are Intimately Linked Rosalina Wisastra and Frank J. Dekker * Pharmaceutical Gene Modulation, Groningen Research Institute of Pharmacy, University of Groningen, Antonius Deusinglaan 1, 9713 AV Groningen, The Netherlands; E-Mail: [email protected] * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +31-5-3638030; Fax: +31-5-3637953. Received: 16 April 2014; in revised form: 27 June 2014 / Accepted: 2 July 2014 / Published: 17 July 2014 Abstract: Cancer and inflammation are intimately linked due to specific oxidative processes in the tumor microenvironment. Lipoxygenases are a versatile class of oxidative enzymes involved in arachidonic acid metabolism. An increasing number of arachidonic acid metabolites is being discovered and apart from their classically recognized pro-inflammatory effects, anti-inflammatory effects are also being described in recent years. Interestingly, these lipid mediators are involved in activation of pro-inflammatory signal transduction pathways such as the nuclear factor κB (NF-κB) pathway, which illustrates the intimate link between lipid signaling and transcription factor activation. The identification of the role of arachidonic acid metabolites in several inflammatory diseases led to a significant drug discovery effort around arachidonic acid metabolizing enzymes. However, to date success in this area has been limited. This might be attributed to the lack of selectivity of the developed inhibitors and to a lack of detailed understanding of the functional roles of arachidonic acid metabolites in inflammatory responses and cancer.