Management of Iron Deficiency Anemia
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Section 8: Hematology CHAPTER 47: ANEMIA
Section 8: Hematology CHAPTER 47: ANEMIA Q.1. A 56-year-old man presents with symptoms of severe dyspnea on exertion and fatigue. His laboratory values are as follows: Hemoglobin 6.0 g/dL (normal: 12–15 g/dL) Hematocrit 18% (normal: 36%–46%) RBC count 2 million/L (normal: 4–5.2 million/L) Reticulocyte count 3% (normal: 0.5%–1.5%) Which of the following caused this man’s anemia? A. Decreased red cell production B. Increased red cell destruction C. Acute blood loss (hemorrhage) D. There is insufficient information to make a determination Answer: A. This man presents with anemia and an elevated reticulocyte count which seems to suggest a hemolytic process. His reticulocyte count, however, has not been corrected for the degree of anemia he displays. This can be done by calculating his corrected reticulocyte count ([3% × (18%/45%)] = 1.2%), which is less than 2 and thus suggestive of a hypoproliferative process (decreased red cell production). Q.2. A 25-year-old man with pancytopenia undergoes bone marrow aspiration and biopsy, which reveals profound hypocellularity and virtual absence of hematopoietic cells. Cytogenetic analysis of the bone marrow does not reveal any abnormalities. Despite red blood cell and platelet transfusions, his pancytopenia worsens. Histocompatibility testing of his only sister fails to reveal a match. What would be the most appropriate course of therapy? A. Antithymocyte globulin, cyclosporine, and prednisone B. Prednisone alone C. Supportive therapy with chronic blood and platelet transfusions only D. Methotrexate and prednisone E. Bone marrow transplant Answer: A. Although supportive care with transfusions is necessary for treating this patient with aplastic anemia, most cases are not self-limited. -

A Distant Gene Deletion Affects Beta-Globin Gene Function in an Atypical Gamma Delta Beta-Thalassemia
A distant gene deletion affects beta-globin gene function in an atypical gamma delta beta-thalassemia. P Curtin, … , A D Stephens, H Lehmann J Clin Invest. 1985;76(4):1554-1558. https://doi.org/10.1172/JCI112136. Research Article We describe an English family with an atypical gamma delta beta-thalassemia syndrome. Heterozygosity results in a beta-thalassemia phenotype with normal hemoglobin A2. However, unlike previously described cases, no history of neonatal hemolytic anemia requiring blood transfusion was obtained. Gene mapping showed a deletion that extended from the third exon of the G gamma-globin gene upstream for approximately 100 kilobases (kb). The A gamma-globin, psi beta-, delta-, and beta-globin genes in cis remained intact. The malfunction of the beta-globin gene on a chromosome in which the deletion is located 25 kb away suggests that chromatin structure and conformation are important for globin gene expression. Find the latest version: https://jci.me/112136/pdf A Distant Gene Deletion Affects ,8-Globin Gene Function in an Atypical '6y5-Thalassemia Peter Curtin, Mario Pirastu, and Yuet Wai Kan Howard Hughes Medical Institute and Department ofMedicine, University of California, San Francisco, California 94143 John Anderson Gobert-Jones Department ofPathology, West Suffolk County Hospital, Bury St. Edmunds IP33-2QZ, Suffolk, England Adrian David Stephens Department ofHaematology, St. Bartholomew's Hospital, London ECIA-7BE, England Herman Lehmann Department ofBiochemistry, University ofCambridge, Cambridge CB2-lQW, England Abstract tologic picture of f3-thalassemia minor in adult life. Globin syn- thetic studies reveal a ,3 to a ratio of -0.5, but unlike the usual We describe an English family with an atypical 'yS6-thalassemia fl-thalassemia heterozygote, the levels of HbA2 (and HbF) are syndrome. -

Anemia in Heart Failure - from Guidelines to Controversies and Challenges
52 Education Anemia in heart failure - from guidelines to controversies and challenges Oana Sîrbu1,*, Mariana Floria1,*, Petru Dascalita*, Alexandra Stoica1,*, Paula Adascalitei, Victorita Sorodoc1,*, Laurentiu Sorodoc1,* *Grigore T. Popa University of Medicine and Pharmacy; Iasi-Romania 1Sf. Spiridon Emergency Hospital; Iasi-Romania ABSTRACT Anemia associated with heart failure is a frequent condition, which may lead to heart function deterioration by the activation of neuro-hormonal mechanisms. Therefore, a vicious circle is present in the relationship of heart failure and anemia. The consequence is reflected upon the pa- tients’ survival, quality of life, and hospital readmissions. Anemia and iron deficiency should be correctly diagnosed and treated in patients with heart failure. The etiology is multifactorial but certainly not fully understood. There is data suggesting that the following factors can cause ane- mia alone or in combination: iron deficiency, inflammation, erythropoietin levels, prescribed medication, hemodilution, and medullar dysfunc- tion. There is data suggesting the association among iron deficiency, inflammation, erythropoietin levels, prescribed medication, hemodilution, and medullar dysfunction. The main pathophysiologic mechanisms, with the strongest evidence-based medicine data, are iron deficiency and inflammation. In clinical practice, the etiology of anemia needs thorough evaluation for determining the best possible therapeutic course. In this context, we must correctly treat the patients’ diseases; according with the current guidelines we have now only one intravenous iron drug. This paper is focused on data about anemia in heart failure, from prevalence to optimal treatment, controversies, and challenges. (Anatol J Cardiol 2018; 20: 52-9) Keywords: anemia, heart failure, intravenous iron, ferric carboxymaltose, quality of life Introduction g/dL in men) (2). -

Eosinophilic Gastroenteritis Presenting with Severe Anemia and Near Syncope
J Am Board Fam Med: first published as 10.3122/jabfm.2012.06.110269 on 7 November 2012. Downloaded from BRIEF REPORT Eosinophilic Gastroenteritis Presenting with Severe Anemia and Near Syncope Nneka Ekunno, DO, MPH, Kirk Munsayac, DO, Allen Pelletier, MD, and Thad Wilkins, MD Eosinophilic gastrointestinal disorders or eosinophilic digestive disorders encompass a spectrum of rare gastrointestinal disorders that includes eosinophilic esophagitis, eosinophilic gastroenteritis, and eosinophilic colitis. Eosinophilic gastroenteritis is a rare inflammatory disease characterized by eosino- philic infiltration of the gastrointestinal tract. The clinical manifestations include anemia, dyspepsia, and diarrhea. Endoscopy with biopsy showing histologic evidence of eosinophilic infiltration is consid- ered definitive for diagnosis. Corticosteroid therapy, food allergen testing, elimination diets, and ele- mental diets are considered effective treatments for eosinophilic gastroenteritis. The treatment and prognosis of eosinophilic gastroenteritis is determined by the severity of the clinical manifestations. We describe a 24-year-old woman with eosinophilic gastroenteritis presenting as epigastric pain with a history of severe iron deficiency anemia, asthma, eczema, and allergic rhinitis, and we review the litera- ture regarding presentation, diagnostic testing, pathophysiology, predisposing factors, and treatment recommendations. (J Am Board Fam Med 2012;25:913–918.) Keywords: Case Reports, Eosinophilic Gastroenteritis, Gastrointestinal Disorders copyright. A 24-year-old nulliparous African-American woman During examination, her height was 62 inches, was admitted after an episode of near syncope asso- weight 117 lb, and body mass index 21.44 kg/m2. Her ciated with 2 days of fatigue and dizziness. She re- heart rate was 111 beats per minute, blood pressure ported gradual onset of dyspepsia over 2 to 3 121/57 mm Hg, respiratory rate 20 breaths per minute, months. -

Alpha Thalassemia Trait
Alpha Thalassemia Trait Alpha Thalassemia Trait Produced by St. Jude Children’s Research Hospital, Departments of Hematology, Patient Education, 1 and Biomedical Communications. Funds were provided by St. Jude Children’s Research Hospital, ALSAC, and a grant from the Plough Foundation. This document is not intended to replace counseling by a trained health care professional or genetic counselor. Our aim is to promote active participation in your care and treatment by providing information and education. Questions about individual health concerns or specific treatment options should be discussed with your doctor. For general information on sickle cell disease and other blood disorders, please visit our Web site at www.stjude.org/sicklecell. Copyright © 2009 St. Jude Children’s Research Hospital Alpha thalassemia trait All red blood cells contain hemoglobin (HEE muh glow bin), which carries oxygen from your lungs to all parts of your body. Alpha thalassemia (thal uh SEE mee uh) trait is a condition that affects the amount of hemo- globin in the red blood cells. • Adult hemoglobin (hemoglobin A) is made of alpha and beta globins. • Normally, people have 4 genes for alpha globin with 2 genes on each chromosome (aa/aa). People with alpha thalassemia trait only have 2 genes for alpha globin, so their bodies make slightly less hemoglobin than normal. This trait was passed on from their parents, like hair color or eye color. A trait is different from a disease 2 Alpha thalassemia trait is not a disease. Normally, a trait will not make you sick. Parents who have alpha thalassemia trait can pass it on to their children. -
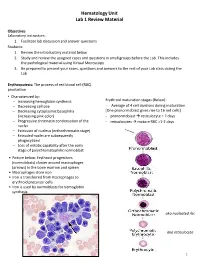
Hematology Unit Lab 1 Review Material
Hematology Unit Lab 1 Review Material Objectives Laboratory instructors: 1. Facilitate lab discussion and answer questions Students: 1. Review the introductory material below 2. Study and review the assigned cases and questions in small groups before the Lab. This includes the pathological material using Virtual Microscopy 3. Be prepared to present your cases, questions and answers to the rest of your Lab class during the Lab Erythropoiesis: The process of red blood cell (RBC) production • Characterized by: − Increasing hemoglobin synthesis Erythroid maturation stages (Below): − Decreasing cell size - Average of 4 cell divisions during maturation − Decreasing cytoplasmic basophilia [One pronormoblast gives rise to 16 red cells] (increasing pink color) - pronormoblast → reticulocyte = 7 days − Progressive chromatin condensation of the - reticulocytes → mature RBC =1-2 days nuclei − Extrusion of nucleus (orthochromatic stage) − Extruded nuclei are subsequently phagocytized − Loss of mitotic capability after the early stage of polychromatophilic normoblast • Picture below: Erythroid progenitors (normoblasts) cluster around macrophages (arrows) in the bone marrow and spleen • Macrophages store iron • Iron is transferred from macrophages to erythroid precursor cells • Iron is used by normoblasts for hemoglobin synthesis aka nucleated rbc aka reticulocyte 1 Mature Red Blood Cell 7-8 microns; round / ovoid biconcave disc with orange-red cytoplasm, no RNA, no nucleus; survives ~120 days in circulation Classification of Anemia by Morphology 1. -

Molecular Epidemiology and Hematologic Characterization of Δβ
Jiang et al. BMC Medical Genetics (2020) 21:43 https://doi.org/10.1186/s12881-020-0981-x RESEARCH ARTICLE Open Access Molecular epidemiology and hematologic characterization of δβ-thalassemia and hereditary persistence of fetal hemoglobin in 125,661 families of greater Guangzhou area, the metropolis of southern China Fan Jiang1,2, Liandong Zuo2, Dongzhi Li2, Jian Li2, Xuewei Tang2, Guilan Chen2, Jianying Zhou2, Hang Lu2 and Can Liao1,2* Abstract Background: Individuals with δβ-thalassemia/HPFH and β-thalassemia usually present with intermedia or thalassemia major. No large-scale survey on HPFH/δβ-thalassemia in southern China has been reported to date. The purpose of this study was to examine the molecular epidemiology and hematologic characteristics of these disorders in Guangzhou, the largest city in Southern China, to offer advice for thalassemia screening programs and genetic counseling. Methods: A total of 125,661 couples participated in pregestational thalassemia screening. 654 subjects with fetal hemoglobin (HbF) level ≥ 5% were selected for further investigation. Gap-PCR combined with Multiplex ligation dependent probe amplification (MLPA) was used to screen for β-globin gene cluster deletions. Gene sequencing for the promoter region of HBG1 /HBG2 gene was performed for all those subjects. Results: A total of 654 individuals had hemoglobin (HbF) levels≥5, and 0.12% of the couples were found to be heterozygous for HPFH/δβ-thalassemia, including Chinese Gγ (Aγδβ)0-thal, Southeast Asia HPFH (SEA-HPFH), Taiwanese deletion and Hb Lepore–Boston–Washington. The highest prevalence was observed in the Huadu district and the lowest in the Nansha district. Three cases were identified as carrying β-globin gene cluster deletions, which had not been previously reported. -

Sickle Cell: It's Your Choice
Sickle Cell: It’s Your Choice What Does “Sickle Cell” Mean? Sickle is a type of hemoglobin. Hemoglobin is the substance that carries oxygen in the blood and gives blood its red color. A person’s hemoglobin type is not the same thing as blood type. The type of hemoglobin we have is determined by genes that we inherit from our parents. The majority of individuals have only the “normal” type of hemoglobin (A). However, there are a variety of other hemoglobin types. Sickle hemoglobin (S) is one of these types. There Are Two Forms of Sickle Cell. Sickle cell occurs in two forms. Sickle cell trait is not a disease; Sickle cell anemia (or sickle cell disease) is a disease. Sickle Cell Trait (or Sickle Trait) Sickle cell trait is found primarily in African Americans, people from areas around the Mediterranean Sea, and from islands in the Caribbean. Sickle cell trait occurs when a person inherits one sickle cell gene from one parent and one normal hemoglobin gene from the other parent. A person with sickle cell trait is healthy and usually is not aware that he or she has the sickle cell gene. A person who has sickle trait can pass it on to their children. If one parent has sickle cell trait and the other parent has the normal type of hemoglobin, there is a 50% (1 in 2) chance with EACH pregnancy that the baby will be born with sickle cell trait. When ONE parent has sickle cell trait, the child may inherit: • 50% chance for two normal hemoglobin genes (normal hemoglobin- AA), OR • 50% chance for one normal hemoglobin gene and one sickle cell gene (sickle cell trait- AS). -

Sickle Cell Disease
Sickle cell disease Description Sickle cell disease is a group of disorders that affects hemoglobin, the molecule in red blood cells that delivers oxygen to cells throughout the body. People with this disease have atypical hemoglobin molecules called hemoglobin S, which can distort red blood cells into a sickle, or crescent, shape. Signs and symptoms of sickle cell disease usually begin in early childhood. Characteristic features of this disorder include a low number of red blood cells (anemia), repeated infections, and periodic episodes of pain. The severity of symptoms varies from person to person. Some people have mild symptoms, while others are frequently hospitalized for more serious complications. The signs and symptoms of sickle cell disease are caused by the sickling of red blood cells. When red blood cells sickle, they break down prematurely, which can lead to anemia. Anemia can cause shortness of breath, fatigue, and delayed growth and development in children. The rapid breakdown of red blood cells may also cause yellowing of the eyes and skin, which are signs of jaundice. Painful episodes can occur when sickled red blood cells, which are stiff and inflexible, get stuck in small blood vessels. These episodes deprive tissues and organs, such as the lungs, kidneys, spleen, and brain, of oxygen-rich blood and can lead to organ damage. A particularly serious complication of sickle cell disease is high blood pressure in the blood vessels that supply the lungs (pulmonary hypertension), which can lead to heart failure. Pulmonary hypertension occurs in about 10 percent of adults with sickle cell disease. Frequency Sickle cell disease affects millions of people worldwide. -

Sickle Cell Disease Brochure
What is sickle cell trait? Who can have sickle cell disease and sickle cell trait? Sickle Cell Trait (AS) is an inherited condition which affects the hemoglobin in your red blood cells. » It is estimated that SCD affects 90,000 to 100,000 people in the United States, mainly Blacks or It is important to know if you have sickle cell trait. African Americans. All About: Sickle cell trait is inherited from your parents, » The disease occurs in about 1 of every 500 Black like hair or eye color. If one parent has sickle cell or African American births and in about 1 of every trait, there is a 50% (1 in 2) chance with each 36,000 Hispanic American births. Sickle Cell pregnancy of having a child with sickle cell trait. Sickle cell trait rarely causes any health problems. » SCD affects millions of people throughout the Some people may develop health problems under world and is particularly common among those certain conditions, such as: whose ancestors come from sub-Saharan Africa, Disease & regions in the Western Hemisphere (South » Dehydration – from not drinking enough water America, the Caribbean, and Central America), » Low oxygen – from over-exertion Saudi Arabia, India, and Mediterranean countries » High altitudes – from low oxygen levels such as Turkey, Greece, and Italy. Sickle Cell » About 1 of every 12 African Americans has sickle How do you know if you have sickle cell cell trait and about 1 of every 100 Hispanics has trait or disease? sickle cell trait. Trait » It is possible for a person of any race or nationality to have sickle cell trait. -

Diagnosis and Management of Autoimmune Hemolytic Anemia in Patients with Liver and Bowel Disorders
Journal of Clinical Medicine Review Diagnosis and Management of Autoimmune Hemolytic Anemia in Patients with Liver and Bowel Disorders Cristiana Bianco 1 , Elena Coluccio 1, Daniele Prati 1 and Luca Valenti 1,2,* 1 Department of Transfusion Medicine and Hematology, Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, 20122 Milan, Italy; [email protected] (C.B.); [email protected] (E.C.); [email protected] (D.P.) 2 Department of Pathophysiology and Transplantation, Università degli Studi di Milano, 20122 Milan, Italy * Correspondence: [email protected]; Tel.: +39-02-50320278; Fax: +39-02-50320296 Abstract: Anemia is a common feature of liver and bowel diseases. Although the main causes of anemia in these conditions are represented by gastrointestinal bleeding and iron deficiency, autoimmune hemolytic anemia should be considered in the differential diagnosis. Due to the epidemiological association, autoimmune hemolytic anemia should particularly be suspected in patients affected by inflammatory and autoimmune diseases, such as autoimmune or acute viral hepatitis, primary biliary cholangitis, and inflammatory bowel disease. In the presence of biochemical indices of hemolysis, the direct antiglobulin test can detect the presence of warm or cold reacting antibodies, allowing for a prompt treatment. Drug-induced, immune-mediated hemolytic anemia should be ruled out. On the other hand, the choice of treatment should consider possible adverse events related to the underlying conditions. Given the adverse impact of anemia on clinical outcomes, maintaining a high clinical suspicion to reach a prompt diagnosis is the key to establishing an adequate treatment. Keywords: autoimmune hemolytic anemia; chronic liver disease; inflammatory bowel disease; Citation: Bianco, C.; Coluccio, E.; autoimmune disease; autoimmune hepatitis; primary biliary cholangitis; treatment; diagnosis Prati, D.; Valenti, L. -

Hemoglobin Bart's and Alpha Thalassemia Fact Sheet
Health Care Provider Hemoglobinopathy Fact Sheet Hemoglobin Bart’s & Alpha Thalassemia Hemoglobin Bart’s is a tetramer of gamma (fetal) globin chains seen during the newborn period. Its presence indicates that one or more of the four genes that produce alpha globin chains are dysfunctional, causing alpha thalassemia. The more alpha genes affected, the more significant the thalassemia and clinical symptoms. Alpha thalassemia occurs in individuals of all ethnic backgrounds and is one of the most common genetic diseases worldwide. However, the clinically significant forms (Hemoglobin H disease, Hemoglobin H Constant Spring, and Alpha Thalassemia Major) occur predominantly among Southeast Asians. Summarized below are the manifestations associated with the different levels of Hemoglobin Bart’s detected on the newborn screen, and recommendations for follow-up. The number of dysfunctional genes is estimated by the percentage of Bart’s seen on the newborn screen. Silent Carrier- Low Bart’s If only one alpha gene is affected, the other three genes can compensate nearly completely and only a low level of Bart’s is detected, unless hemoglobin Constant Spring is identified (see below). Levels of Bart’s below a certain percentage are not generally reported by the State Newborn Screening Program as these individuals are likely to be clinically and hematologically normal. However, a small number of babies reported as having possible alpha thalassemia trait will be silent carriers. Alpha Thalassemia or Hemoglobin Constant Spring Trait- Moderate Bart’s Alpha thalassemia trait produces a moderate level of Bart’s and typically results from the dysfunction of two alpha genes-- either due to gene deletions or a specific change in the alpha gene that produces elongated alpha globin and has a thalassemia-like effect: hemoglobin Constant Spring.