A Distant Gene Deletion Affects Beta-Globin Gene Function in an Atypical Gamma Delta Beta-Thalassemia
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Developmental Genetics of Hemoglobin Synthesis in Chironomus Darrel Starr English Iowa State University
Iowa State University Capstones, Theses and Retrospective Theses and Dissertations Dissertations 1968 The developmental genetics of hemoglobin synthesis in Chironomus Darrel Starr English Iowa State University Follow this and additional works at: https://lib.dr.iastate.edu/rtd Part of the Genetics Commons Recommended Citation English, Darrel Starr, "The developmental genetics of hemoglobin synthesis in Chironomus " (1968). Retrospective Theses and Dissertations. 3660. https://lib.dr.iastate.edu/rtd/3660 This Dissertation is brought to you for free and open access by the Iowa State University Capstones, Theses and Dissertations at Iowa State University Digital Repository. It has been accepted for inclusion in Retrospective Theses and Dissertations by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. This dissertation has been microfilmed exactly as received 6 8-14,785 ENGLISH, Barrel Starr, 1936- THE DEVELOPMENTAL GENETICS OF HEMOGLOBIN SYNTHESIS IN CHIRONOMUS. Iowa State University, Ph.D., 1968 Biology- Genetics University Microfilms, Inc., Ann Arbor, Michigan THE DEVELOPMENTAL GENETICS OF HEMOGLOBIN SYNTHESIS IN CHIRONOMUS by Darrel Starr English A Dissertation Submitted to the Graduate Faculty in Partial Fulfillment of The Requirements for the Degree of DOCTOR OF PHILOSOPHY Major Subject: Genetics Approved: Signature was redacted for privacy. In Charge of MajdA Work Signature was redacted for privacy. Head ^ Major Department Signature was redacted for privacy. -

Section 8: Hematology CHAPTER 47: ANEMIA
Section 8: Hematology CHAPTER 47: ANEMIA Q.1. A 56-year-old man presents with symptoms of severe dyspnea on exertion and fatigue. His laboratory values are as follows: Hemoglobin 6.0 g/dL (normal: 12–15 g/dL) Hematocrit 18% (normal: 36%–46%) RBC count 2 million/L (normal: 4–5.2 million/L) Reticulocyte count 3% (normal: 0.5%–1.5%) Which of the following caused this man’s anemia? A. Decreased red cell production B. Increased red cell destruction C. Acute blood loss (hemorrhage) D. There is insufficient information to make a determination Answer: A. This man presents with anemia and an elevated reticulocyte count which seems to suggest a hemolytic process. His reticulocyte count, however, has not been corrected for the degree of anemia he displays. This can be done by calculating his corrected reticulocyte count ([3% × (18%/45%)] = 1.2%), which is less than 2 and thus suggestive of a hypoproliferative process (decreased red cell production). Q.2. A 25-year-old man with pancytopenia undergoes bone marrow aspiration and biopsy, which reveals profound hypocellularity and virtual absence of hematopoietic cells. Cytogenetic analysis of the bone marrow does not reveal any abnormalities. Despite red blood cell and platelet transfusions, his pancytopenia worsens. Histocompatibility testing of his only sister fails to reveal a match. What would be the most appropriate course of therapy? A. Antithymocyte globulin, cyclosporine, and prednisone B. Prednisone alone C. Supportive therapy with chronic blood and platelet transfusions only D. Methotrexate and prednisone E. Bone marrow transplant Answer: A. Although supportive care with transfusions is necessary for treating this patient with aplastic anemia, most cases are not self-limited. -

8.5 X12.5 Doublelines.P65
Cambridge University Press 978-0-521-87519-6 - Disorders of Hemoglobin: Genetics, Pathophysiology, and Clinical Management, Second Edition Edited by Martin H. Steinberg, Bernard G. Forget, Douglas R. Higgs and David J. Weatherall Index More information anti-inflammatory therapy, 762–763 thalassemia-related complications, 779 sulfasalazine, nuclear factor (NF)-kB, 762 transplant-related complications, 778–779 targeting ET-1, 762–763 S-linked haplotypes, African/Indo-European, anti-oxidant therapy targeting erythrocyte, 638–640 765–766 burst forming unit-erythroid (BFU-E), 10, 29 deferiprone, 765 oral glutamine, 765 calcium-activated potassium channel (Gardos oral N-acetyl-cysteine, 765–766 pathway), 167–168 anti-oxidant therapy targeting vascular, 763–765 capillary electrophoresis, 660 Index Apo A-I mimetics, 764 capillary IEF, 660 NO, 763–764 carbon monoxide poisoning, 613–616 statins, 764 clinical features, 615 xanthine oxidase inhibitors, 764–765 diagnosis, treatment, 615–616 anti-thrombotic therapy epidemiology, 613–614 -thalassemia, 761–762 cardiac, arterial abnormalities, 151 sickle cell disease, 761–762 cardiac abnormalities, ATRX syndrome, 305 aortagonad-mesonephros (AGM), 6 cardiovascular disease, 652 Apo A-I mimetics, 764 cation content, cellular hydration, 164–172 apoptosis, vasculature permeability, 193–194 calcium-activated potassium channel, 167–168 assays, assay systems, 7, 142 cation permeability alterations, 166–167 ATMDS. See ␣ thalassemia-myelodysplastic cell calcium, calcium pump activity, 167 syndromes cell sodium, -

Anemia in Heart Failure - from Guidelines to Controversies and Challenges
52 Education Anemia in heart failure - from guidelines to controversies and challenges Oana Sîrbu1,*, Mariana Floria1,*, Petru Dascalita*, Alexandra Stoica1,*, Paula Adascalitei, Victorita Sorodoc1,*, Laurentiu Sorodoc1,* *Grigore T. Popa University of Medicine and Pharmacy; Iasi-Romania 1Sf. Spiridon Emergency Hospital; Iasi-Romania ABSTRACT Anemia associated with heart failure is a frequent condition, which may lead to heart function deterioration by the activation of neuro-hormonal mechanisms. Therefore, a vicious circle is present in the relationship of heart failure and anemia. The consequence is reflected upon the pa- tients’ survival, quality of life, and hospital readmissions. Anemia and iron deficiency should be correctly diagnosed and treated in patients with heart failure. The etiology is multifactorial but certainly not fully understood. There is data suggesting that the following factors can cause ane- mia alone or in combination: iron deficiency, inflammation, erythropoietin levels, prescribed medication, hemodilution, and medullar dysfunc- tion. There is data suggesting the association among iron deficiency, inflammation, erythropoietin levels, prescribed medication, hemodilution, and medullar dysfunction. The main pathophysiologic mechanisms, with the strongest evidence-based medicine data, are iron deficiency and inflammation. In clinical practice, the etiology of anemia needs thorough evaluation for determining the best possible therapeutic course. In this context, we must correctly treat the patients’ diseases; according with the current guidelines we have now only one intravenous iron drug. This paper is focused on data about anemia in heart failure, from prevalence to optimal treatment, controversies, and challenges. (Anatol J Cardiol 2018; 20: 52-9) Keywords: anemia, heart failure, intravenous iron, ferric carboxymaltose, quality of life Introduction g/dL in men) (2). -

Eosinophilic Gastroenteritis Presenting with Severe Anemia and Near Syncope
J Am Board Fam Med: first published as 10.3122/jabfm.2012.06.110269 on 7 November 2012. Downloaded from BRIEF REPORT Eosinophilic Gastroenteritis Presenting with Severe Anemia and Near Syncope Nneka Ekunno, DO, MPH, Kirk Munsayac, DO, Allen Pelletier, MD, and Thad Wilkins, MD Eosinophilic gastrointestinal disorders or eosinophilic digestive disorders encompass a spectrum of rare gastrointestinal disorders that includes eosinophilic esophagitis, eosinophilic gastroenteritis, and eosinophilic colitis. Eosinophilic gastroenteritis is a rare inflammatory disease characterized by eosino- philic infiltration of the gastrointestinal tract. The clinical manifestations include anemia, dyspepsia, and diarrhea. Endoscopy with biopsy showing histologic evidence of eosinophilic infiltration is consid- ered definitive for diagnosis. Corticosteroid therapy, food allergen testing, elimination diets, and ele- mental diets are considered effective treatments for eosinophilic gastroenteritis. The treatment and prognosis of eosinophilic gastroenteritis is determined by the severity of the clinical manifestations. We describe a 24-year-old woman with eosinophilic gastroenteritis presenting as epigastric pain with a history of severe iron deficiency anemia, asthma, eczema, and allergic rhinitis, and we review the litera- ture regarding presentation, diagnostic testing, pathophysiology, predisposing factors, and treatment recommendations. (J Am Board Fam Med 2012;25:913–918.) Keywords: Case Reports, Eosinophilic Gastroenteritis, Gastrointestinal Disorders copyright. A 24-year-old nulliparous African-American woman During examination, her height was 62 inches, was admitted after an episode of near syncope asso- weight 117 lb, and body mass index 21.44 kg/m2. Her ciated with 2 days of fatigue and dizziness. She re- heart rate was 111 beats per minute, blood pressure ported gradual onset of dyspepsia over 2 to 3 121/57 mm Hg, respiratory rate 20 breaths per minute, months. -

Alpha Thalassemia Trait
Alpha Thalassemia Trait Alpha Thalassemia Trait Produced by St. Jude Children’s Research Hospital, Departments of Hematology, Patient Education, 1 and Biomedical Communications. Funds were provided by St. Jude Children’s Research Hospital, ALSAC, and a grant from the Plough Foundation. This document is not intended to replace counseling by a trained health care professional or genetic counselor. Our aim is to promote active participation in your care and treatment by providing information and education. Questions about individual health concerns or specific treatment options should be discussed with your doctor. For general information on sickle cell disease and other blood disorders, please visit our Web site at www.stjude.org/sicklecell. Copyright © 2009 St. Jude Children’s Research Hospital Alpha thalassemia trait All red blood cells contain hemoglobin (HEE muh glow bin), which carries oxygen from your lungs to all parts of your body. Alpha thalassemia (thal uh SEE mee uh) trait is a condition that affects the amount of hemo- globin in the red blood cells. • Adult hemoglobin (hemoglobin A) is made of alpha and beta globins. • Normally, people have 4 genes for alpha globin with 2 genes on each chromosome (aa/aa). People with alpha thalassemia trait only have 2 genes for alpha globin, so their bodies make slightly less hemoglobin than normal. This trait was passed on from their parents, like hair color or eye color. A trait is different from a disease 2 Alpha thalassemia trait is not a disease. Normally, a trait will not make you sick. Parents who have alpha thalassemia trait can pass it on to their children. -
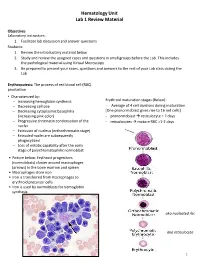
Hematology Unit Lab 1 Review Material
Hematology Unit Lab 1 Review Material Objectives Laboratory instructors: 1. Facilitate lab discussion and answer questions Students: 1. Review the introductory material below 2. Study and review the assigned cases and questions in small groups before the Lab. This includes the pathological material using Virtual Microscopy 3. Be prepared to present your cases, questions and answers to the rest of your Lab class during the Lab Erythropoiesis: The process of red blood cell (RBC) production • Characterized by: − Increasing hemoglobin synthesis Erythroid maturation stages (Below): − Decreasing cell size - Average of 4 cell divisions during maturation − Decreasing cytoplasmic basophilia [One pronormoblast gives rise to 16 red cells] (increasing pink color) - pronormoblast → reticulocyte = 7 days − Progressive chromatin condensation of the - reticulocytes → mature RBC =1-2 days nuclei − Extrusion of nucleus (orthochromatic stage) − Extruded nuclei are subsequently phagocytized − Loss of mitotic capability after the early stage of polychromatophilic normoblast • Picture below: Erythroid progenitors (normoblasts) cluster around macrophages (arrows) in the bone marrow and spleen • Macrophages store iron • Iron is transferred from macrophages to erythroid precursor cells • Iron is used by normoblasts for hemoglobin synthesis aka nucleated rbc aka reticulocyte 1 Mature Red Blood Cell 7-8 microns; round / ovoid biconcave disc with orange-red cytoplasm, no RNA, no nucleus; survives ~120 days in circulation Classification of Anemia by Morphology 1. -

Molecular Epidemiology and Hematologic Characterization of Δβ
Jiang et al. BMC Medical Genetics (2020) 21:43 https://doi.org/10.1186/s12881-020-0981-x RESEARCH ARTICLE Open Access Molecular epidemiology and hematologic characterization of δβ-thalassemia and hereditary persistence of fetal hemoglobin in 125,661 families of greater Guangzhou area, the metropolis of southern China Fan Jiang1,2, Liandong Zuo2, Dongzhi Li2, Jian Li2, Xuewei Tang2, Guilan Chen2, Jianying Zhou2, Hang Lu2 and Can Liao1,2* Abstract Background: Individuals with δβ-thalassemia/HPFH and β-thalassemia usually present with intermedia or thalassemia major. No large-scale survey on HPFH/δβ-thalassemia in southern China has been reported to date. The purpose of this study was to examine the molecular epidemiology and hematologic characteristics of these disorders in Guangzhou, the largest city in Southern China, to offer advice for thalassemia screening programs and genetic counseling. Methods: A total of 125,661 couples participated in pregestational thalassemia screening. 654 subjects with fetal hemoglobin (HbF) level ≥ 5% were selected for further investigation. Gap-PCR combined with Multiplex ligation dependent probe amplification (MLPA) was used to screen for β-globin gene cluster deletions. Gene sequencing for the promoter region of HBG1 /HBG2 gene was performed for all those subjects. Results: A total of 654 individuals had hemoglobin (HbF) levels≥5, and 0.12% of the couples were found to be heterozygous for HPFH/δβ-thalassemia, including Chinese Gγ (Aγδβ)0-thal, Southeast Asia HPFH (SEA-HPFH), Taiwanese deletion and Hb Lepore–Boston–Washington. The highest prevalence was observed in the Huadu district and the lowest in the Nansha district. Three cases were identified as carrying β-globin gene cluster deletions, which had not been previously reported. -

Oxygen Equilibria of Hemoglobin A2 and Hemoglobin Lepore
Oxygen Equilibria of Hemoglobin A2 and Hemoglobin Lepore Grace G. Eddison, … , Robin W. Briehl, Helen M. Ranney J Clin Invest. 1964;43(12):2323-2331. https://doi.org/10.1172/JCI105106. Research Article Find the latest version: https://jci.me/105106/pdf Journal of Clinical Investigation Vol. 43, No. 12, 1964 Oxygen Equilibria of Hemoglobin A2 and Hemoglobin Lepore * GRACE G. EDDISON,t ROBIN W. BRIEHL,$ AND HELEN M. RANNEY (From the Departments of Medicine and Physiology of the Albert Einstein College of Medi- cine and the Bronx Municipal Hospital Center, New York, N. Y.) Human hemoglobin provides a model for studies the oxygen equilibria of erythrocytes obtained concerned with the relationships of structure and from an adult in whom hemoglobin F comprised biologic function of proteins. Older evidence for 69%o of the total pigment resembled the oxygen conformational differences between oxygenated equilibria of normal adult blood rather than that and deoxygenated normal hemoglobin (1) has of cord blood. These workers (8) suggested that recently been confirmed and extended by X-ray differences between the fetal and adult red cell crystallographic studies (2) and by comparison of other than the type of hemoglobin must be con- the dissociation (3) and of the hybridization (4) cerned in the oxygenation function of whole blood of the oxygenated and deoxygenated pigments. obtained from newborn infants. Normal and abnormal human hemoglobins have Although hemolysates containing large pro- been utilized in the past by other workers for in- portions of hemoglobins F or A can be studied di- vestigation of relationships between structure and rectly, isolation procedures must be utilized to oxygen equilibria. -

Regulations Governing the Classification of Medical Devices (Draft)
Regulations Governing the Classification of Medical Devices (Draft) Article 1 These Regulations are enacted pursuant to Paragraph 2, Article 3 of the Medical Devices Act (hereinafter “this Act”). Article 2 Medical devices are classified into the following categories according to their function, intended use, operating instructions, and working principle, depending on the applicable medical specialty: 1. Clinical chemistry and clinical toxicology devices 2. Hematology and pathology devices 3. Immunology and microbiology devices 4. Anesthesiology devices 5. Cardiovascular devices 6. Dental devices 7. Ear, nose, and throat devices 8. Gastroenterology and urology devices 9. General and plastic surgery devices 10. General hospital and personal use devices 11. Neurological devices 12. Obstetrical and gynecological devices 13. Ophthalmic devices 14. Orthopedic devices 15. Physical medicine devices 16. Radiology devices Article 3 Medical devices are classified into the following classes according to their risk level: 1. Class I: Low risk 2. Class II: Medium risk 3. Class III: High risk 1 Article 4 Product items of the medical device classification are specified in the Annex. In addition to rules stated in the Annex, medical devices whose function, intended use, or working principle are special may have their classification determined according to the following rules: 1. If two or more categories, classes, or product items are applicable to the same medical device, the highest class of risk level is assigned. 2. The accessory to a medical device, intended specifically by the manufacturer for use with a particular medical device, is classified the same as the particular medical device, unless otherwise specified in the Annex. 3. -

Sickle Cell: It's Your Choice
Sickle Cell: It’s Your Choice What Does “Sickle Cell” Mean? Sickle is a type of hemoglobin. Hemoglobin is the substance that carries oxygen in the blood and gives blood its red color. A person’s hemoglobin type is not the same thing as blood type. The type of hemoglobin we have is determined by genes that we inherit from our parents. The majority of individuals have only the “normal” type of hemoglobin (A). However, there are a variety of other hemoglobin types. Sickle hemoglobin (S) is one of these types. There Are Two Forms of Sickle Cell. Sickle cell occurs in two forms. Sickle cell trait is not a disease; Sickle cell anemia (or sickle cell disease) is a disease. Sickle Cell Trait (or Sickle Trait) Sickle cell trait is found primarily in African Americans, people from areas around the Mediterranean Sea, and from islands in the Caribbean. Sickle cell trait occurs when a person inherits one sickle cell gene from one parent and one normal hemoglobin gene from the other parent. A person with sickle cell trait is healthy and usually is not aware that he or she has the sickle cell gene. A person who has sickle trait can pass it on to their children. If one parent has sickle cell trait and the other parent has the normal type of hemoglobin, there is a 50% (1 in 2) chance with EACH pregnancy that the baby will be born with sickle cell trait. When ONE parent has sickle cell trait, the child may inherit: • 50% chance for two normal hemoglobin genes (normal hemoglobin- AA), OR • 50% chance for one normal hemoglobin gene and one sickle cell gene (sickle cell trait- AS). -

Hematology Notes Blood/ Hematology Danil Hammoudi.MD
Hematology notes Blood/ Hematology Danil Hammoudi.MD HTTP://Sinoemedicalassociation.or/AP2/ Page | 1 Blood is a connective tissue whose matrix is fluid. It is composed of: 1. red corpuscles, 2. white cells, 3. platelets, 4. blood plasma. It is transported throughout the body within blood vessels. • Blood is sometimes considered to be a fluid connective tissue because of the mesenchymal origin of its cells and a low ratio of cells to liquid intercellular substance, the blood plasma. • In human adults about 5 liter of blood contribute 7-8 % to the body weight of the individual. • The contribution of red blood cells (erythrocytes) to the total volume of the blood (haematocrit) is about 43%. • Erythrocytes are the dominant (99%) but not the only type of cells in the blood. • We also find leukocytes and, in addition, blood platelets. Erythrocytes, leukocytes and blood platelets are also being referred to as the formed elements of the blood. • Erythrocytes and blood platelets perform their functions exclusively in the blood stream. • In contrast, leukocytes reside only temporarily in the blood. • Leukocytes can leave the blood stream through the walls of capillaries and venules and enter either connective or lymphoid tissues. Hematology notes Page | 2 Hematology notes Page | 3 Blood facts • Approximately 8% of an adult's body weight is made up of blood. • Females have around 4-5 litres, while males have around 5-6 litres. This difference is mainly due to the differences in body size between men and women. • Its mean temperature is 38 degrees Celcius. • It has a pH of 7.35-7.45, making it slightly basic (less than 7 is considered acidic).