Phylogenetic Analysis of Different Ploidy Saccharum Spontaneum Based on Rdna-ITS Sequences
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Sugarcane (Saccharum Officinarum L.)
/4 ^'^ SUGARCANE (SACCHARUM OFFICINARUM L.) ORIGIN CLASSIFICATION CHARACTERISTICS, AND DESCRIPTIONS OF REPRESENTATIVE CLONES ■ u AGRICULTURE HANDBOOK NO. 122 UNITED STATES DEPARTMENT OF AGRICULTURE SUGARCANE (SACCHARUM OFFICINARUM L.) ORIGIN CLASSIFICATION CHARACTERISTICS AND DESCRIPTIONS OF REPRESENTATIVE CLONES By Ernst Artschwager Formerly Senior Botanist and E. W. Brandes Formerly Head Pathologist Crops Research Division Agricultural Research Service AGRICULTURE HANDBOOK No. 122 The garden sugarcanes of Melanesia constitute the original base from which our present-day varieties of sugarcane derive. Varietal descriptions of the Melanesian garden canes and their derivatives are drawn from a living collection and are here placed on record. As background material and to promote fullest interest in use of this extensive store of germ plasm, what is known or what logically may be in- ferred concerning the history, value, and use of these varieties as a group and their interrelationships with other varietal groups in the genus is given. CONTENTS Page Page Origin, classification, and charac- Vegetative characters used in the teristics 1 description and classification Importance of Saccharum offici- of noble canes—Continued narum 3 Vegetative characters—Con. Colloquial names 3 Blade 56 Relative position of Saccharum Leaf sheath 57 officinarum in the genus 7 Auricles 59 Geographic origin and dispersal. 18 Ligule 61 Origin 18 Dewlaps 61 Dispersal 21 Midrib pubescence 64 Collecting expeditions, 1853- Evaluation of characters used_- 65 1951 28 Descriptions and taxonomic keys Vegetative characters used in the of the clones 81 description and classification Descriptions of the clones 83 of noble canes 45 New Guinea group 83 Materials and methods 45 New Caledonian group 160 Vegetative characters 48 Hawaiian group 180 v Internode 48 Miscellaneous noble group 198 Bud 52 Taxonomic keys 253 Leaf 56 Literature cited 260 Washington, D. -

Operation China
Lipo, Western July 4 Location: More than 146,000 past. While the Western Lipo live in an impoverished Eastern Lipo are a area of northern Yunnan Province. Lisu-speaking group They inhabit six counties in Yunnan — who migrated to Dayao, Yongren, Yuanmou, Binchuan, their present Yao’an, and Yongsheng. A small location after a number of Western Lipo spill across military defeat in the border into the Renhe District of 1812,3 the Western Panzhihua County in Sichuan Province. Lipo claim to have originated in Nanjing Identity: There is a great deal of or Jiangxi in eastern confusion regarding the classification China. According to of the Lipo. The Lipo as a whole have local accounts, “the been officially included as part of the ancestors of today’s Yi nationality in China, yet among the Lipuo [Western Lipo are two very distinct groups, Lipo] came as which here are defined as Eastern soldiers at various and Western Lipo. The two do not times from the early consider themselves to be the same Ming to the early people. They have different histories, Qing dynasties [late dress, customs, and languages. This 14th century to late century a further distinction has 17th century]. developed between the two groups: Historical records the Eastern Lipo are known as a confirm that those Christian tribe while the Western Lipo dynasties did send Jamin Pelkey have relatively few Christians. Lipo military expeditions to the area that is Religion: The majority of Western Lipo means “insiders.” They refer to the now Yongren; we can speculate that are polytheists. They worship many Han Chinese as Xipo, or “outsiders.” today’s Lipuo [Western Lipo] are spirits and protective deities, including descendants of intermarriage between mountain deities. -

Operation China Their Alternate the Three Main Religions Among the Tusu Name Is Used to Avoid Confusion with the People
Tusu November 27 of Western Yi. Tusu speakers, SICHUAN •Lijiang however, cannot understand the speech of other Western Yi •Binchuan •Dayao •Dali Kunming groups in their region and • •Chuxiong must revert to Chinese in •Yunxian •Wanding order to communicate. YUNNAN Scale 0 KM 160 History: For more than 1,300 Population in China: years the Tusu have appeared 31,000 (1999) in Chinese records of Yunnan 31,750 (2000) Province. The Tusu “are said 39,900 (2010) Location: Yunnan to be descendants of the Religion: Polytheism ancient Muocha tribe which Christians: 20 moved from Baoshan to Weishan in ancient times. In Overview of the Tusu the seventh century AD they Countries: China moved to present-day Xiangyun Pronunciation: “Too-soo” County and later spread into Other Names: southeastern Dayao and parts 2 Tusupo, Tu, Turen, Tuzu of Binchuan County.” Population Source: 31,000 (1999 J. Pelkey); Customs: In some areas the Out of a total Yi population of 6,572,173 (1990 census) Tusu love to come together Location: N Yunnan: Xiangyun and participate in local (15,000), Binchuan (13,000), festivals, since it gives them a and Dayao (3,000) counties Jamin Pelkey chance to relax and forget Status: Location: Approximately 31,000 Tusu about their hardships and struggles. The Officially included under Yi people live in the western central part of festivals also serve as a reunion for Language: Sino-Tibetan, Tibeto-Burman, Burmese-Lolo, Yunnan Province in southwest China. In relatives and friends. The Tiger Dance Lolo, Northern Lolo, Yi, Xiangyun County the Tusu inhabit the Festival is held for one week during the first Western Yi Da’aonai Community of Luwu District; the lunar month each year. -
A Revision of the Genus Pseudoechthistatus Pic
A peer-reviewed open-access journal ZooKeys 604: 49–85 (2016) A revision of the genus Pseudoechthistatus Pic... 49 doi: 10.3897/zookeys.604.9049 RESEARCH ARTICLE http://zookeys.pensoft.net Launched to accelerate biodiversity research A revision of the genus Pseudoechthistatus Pic (Coleoptera, Cerambycidae, Lamiinae, Lamiini) Wen-Xuan Bi1,2, Mei-Ying Lin1 1 Key Laboratory of Zoological Systematics and Evolution, Institute of Zoology, Chinese Academy of Sciences, Beichen West Road, Chaoyang, Beijing, 100101, China 2 Room 401, No. 2, Lane 155, Lianhua South Road, Shanghai, 201100, China Corresponding authors: Wen-Xuan Bi ([email protected]); Mei-Ying Lin ([email protected]) Academic editor: F. Vitali | Received 29 April 2016 | Accepted 19 May 2016 | Published 11 July 2016 http://zoobank.org/28522BEE-2F2A-4E8B-A0B3-5FB901671E85 Citation: Bi W-X, Lin M-Y (2016) A revision of the genus Pseudoechthistatus Pic (Coleoptera, Cerambycidae, Lamiinae, Lamiini). ZooKeys 604: 49–85. doi: 10.3897/zookeys.604.9049 Abstract The genus Pseudoechthistatus Pic, 1917 is redefined and revised. Five species of the genus are described as new, P. sinicus sp. n. and P. chiangshunani sp. n. from central Yunnan, China, P. pufujiae sp. n. from west- ern Yunnan, China, and P. holzschuhi sp. n. and P. glabripennis sp. n. from southern Yunnan and northern Vietnam. Pseudoechthistatus birmanicus Breuning, 1942 is excluded from the fauna of China. Three poorly known species, P. obliquefasciatus Pic, 1917, P. granulatus Breuning, 1942, and P. acutipennis Chiang, 1981 are redescribed, and the type localities of the former two species are discussed. Endophallic structure of seven species in inflated and everted condition are studied and compared with their relatives. -

Ecological Indicators Regional Ecosystem Health Response to Rural
Ecological Indicators 72 (2017) 399–410 Contents lists available at ScienceDirect Ecological Indicators j ournal homepage: www.elsevier.com/locate/ecolind Original Articles Regional ecosystem health response to rural land use change: A case study in Lijiang City, China a,∗ a a a,b Peng Jian , Liu Yanxu , Li Tianyi , Wu Jiansheng a Laboratory for Earth Surface Processes, Ministry of Education, College of Urban and Environmental Sciences, Peking University, Beijing 100871, China b Key Laboratory for Environmental and Urban Sciences, School of Urban Planning and Design, Shenzhen Graduate School, Peking University, Shenzhen 518055, China a r t i c l e i n f o a b s t r a c t Article history: Quantitative analysis of the response of ecosystem health to rural land use change is required to compre- Received 22 December 2015 hend the human-nature coupling mechanism and to explore the process of global environmental change, Received in revised form 13 August 2016 which can interpret the ecological effects of regional land use and land cover change comprehensively. Accepted 16 August 2016 However, the existing regional ecosystem health assessment largely ignored either the internal connec- Available online 3 September 2016 tion of ecosystem health to land use patterns or the internal representation of ecosystem services to ecosystem health. Using Lijiang City of China as a study area, the average normalized difference vege- Keywords: tation index (NDVI), landscape metrics, and ecosystem elasticity coefficient based on different land use Regional ecosystem health assessment types were used as quantitative indicators. Then the coefficient of spatial neighboring effect was intro- Ecosystem services duced to characterize the adjacency effect on ecosystem services, and to generate the index of integrated Land use change Lijiang City, China ecosystem health. -
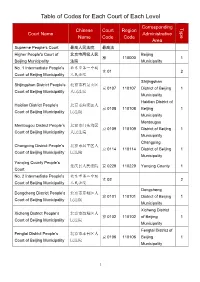
Table of Codes for Each Court of Each Level
Table of Codes for Each Court of Each Level Corresponding Type Chinese Court Region Court Name Administrative Name Code Code Area Supreme People’s Court 最高人民法院 最高法 Higher People's Court of 北京市高级人民 Beijing 京 110000 1 Beijing Municipality 法院 Municipality No. 1 Intermediate People's 北京市第一中级 京 01 2 Court of Beijing Municipality 人民法院 Shijingshan Shijingshan District People’s 北京市石景山区 京 0107 110107 District of Beijing 1 Court of Beijing Municipality 人民法院 Municipality Haidian District of Haidian District People’s 北京市海淀区人 京 0108 110108 Beijing 1 Court of Beijing Municipality 民法院 Municipality Mentougou Mentougou District People’s 北京市门头沟区 京 0109 110109 District of Beijing 1 Court of Beijing Municipality 人民法院 Municipality Changping Changping District People’s 北京市昌平区人 京 0114 110114 District of Beijing 1 Court of Beijing Municipality 民法院 Municipality Yanqing County People’s 延庆县人民法院 京 0229 110229 Yanqing County 1 Court No. 2 Intermediate People's 北京市第二中级 京 02 2 Court of Beijing Municipality 人民法院 Dongcheng Dongcheng District People’s 北京市东城区人 京 0101 110101 District of Beijing 1 Court of Beijing Municipality 民法院 Municipality Xicheng District Xicheng District People’s 北京市西城区人 京 0102 110102 of Beijing 1 Court of Beijing Municipality 民法院 Municipality Fengtai District of Fengtai District People’s 北京市丰台区人 京 0106 110106 Beijing 1 Court of Beijing Municipality 民法院 Municipality 1 Fangshan District Fangshan District People’s 北京市房山区人 京 0111 110111 of Beijing 1 Court of Beijing Municipality 民法院 Municipality Daxing District of Daxing District People’s 北京市大兴区人 京 0115 -

Characterization of Chromosome Composition of Sugarcane in Nobilization by Using Genomic in Situ Hybridization
Yu et al. Molecular Cytogenetics (2018) 11:35 https://doi.org/10.1186/s13039-018-0387-z RESEARCH Open Access Characterization of chromosome composition of sugarcane in nobilization by using genomic in situ hybridization Fan Yu1, Ping Wang1, Xueting Li1, Yongji Huang1, Qinnan Wang2, Ling Luo1, Yanfen Jing3, Xinlong Liu3, Zuhu Deng1,4*, Jiayun Wu2, Yongqing Yang1, Rukai Chen1, Muqing Zhang4 and Liangnian Xu1* Abstract Background: Interspecific hybridization is an effective strategy for germplasm innovation in sugarcane. Nobilization refers to the breeding theory of development and utilization of wild germplasm. Saccharum spontaneum is the main donor of resistance and adaptive genes in the nobilization breeding process. Chromosome transfer in sugarcane is complicated; thus, research of different inheritance patterns can provide guidance for optimal sugarcane breeding. Results: Through chromosome counting and genomic in situ hybridization, we found that six clones with 80 chromosomes were typical S. officinarum and four other clones with more than 80 chromosomes were interspecific hybrids between S. officinarum and S. spontaneum. These data support the classical view that S. officinarum is characterized by 2n = 80. In addition, genomic in situ hybridization showed that five F1 clones were products of a 2n + n transmission and one F1 clone was the product of an n + n transmission in clear pedigree noble hybrids between S. officinarum and S. spontaneum. Interestingly, Yacheng 75–408 and Yacheng 75–409 were the sibling lines of the F1 progeny from the same parents but with different genetic transmissions. Conclusions: This is the first clear evidence of Loethers, Crystallina, Luohanzhe, Vietnam Niuzhe, and Nanjian Guozhe were typical S. -

World Bank Documents
Public Disclosure Copy The World Bank Implementation Status & Results Report Yunnan Urban Environment Project (P096812) Yunnan Urban Environment Project (P096812) EAST ASIA AND PACIFIC | China | Water Global Practice | IBRD/IDA | Specific Investment Loan | FY 2009 | Seq No: 9 | ARCHIVED on 21-Apr-2015 | ISR18896 | Public Disclosure Authorized Implementing Agencies: Yunnan Provincial Environment Protection Bureau - Foreign Financed PMO Key Dates Key Project Dates Board Approval date:05-May-2009 Effectiveness Date:30-Sep-2009 Planned Mid Term Review Date:31-May-2013 Actual Mid-Term Review Date:15-Dec-2013 Original Closing Date:30-Jun-2015 Revised Closing Date:30-Jun-2016 Public Disclosure Authorized Project Development Objectives Project Development Objective (from Project Appraisal Document) To assist Yunnan Province in improving the effectiveness and coverage of critical urban infrastructure services in selectedcountiesand the effectiveness of lake basin management in Dianchi, through investment in systems for the management of wastewater,water supply, solid waste, river environment and cultural heritage. Has the Project Development Objective been changed since Board Approval of the Project Objective? No PHRPDODEL Public Disclosure Authorized Components Name Urban Environment Infrastructure Development:(Cost $166.50 M) Integrated Lake Basin Management:(Cost $11.80 M) Water Supply for Zhaotong Municipality:(Cost $49.58 M) Wastewater for Zhaotong Municipality:(Cost $17.21 M) River Rehabilitation for Zhaotong Municipality:(Cost $60.89 M) -

Simulating the Impact of Grain-For-Green Programme on Ecosystem Services Trade-Offs in North-Western Yunnan, China
Simulating the impact of Grain-for-Green Programme on ecosystem services trade-offs in North-western Yunnan, China Jian Penga, b , Xiaoxu Hua, Xiaoyu Wanga, b, Jeroen Meersmansc, Yanxu Liud, Sijing a Qiu a Laboratory for Earth Surface Processes, Ministry of Education, College of Urban and Environmental Sciences, Peking University, Beijing 100871, China b Key Laboratory for Environmental and Urban Sciences, School of Urban Planning and Design, Shenzhen Graduate School, Peking University, Shenzhen 518055, Chinac, Cranfield Soil and Agrifood Institute, School of Water, Energy and Environment, Cranfield University, Cranfield, MK43 0AL, United Kingdom d State Key Laboratory of Earth Surface Processes and Resource Ecology, Faculty of Geographical Science, Beijing Normal University, Beijing 100875, China Abstract: One of the main manifestations of the Grain-for-Green Programme (GFGP) is land use change, which will affect the trade-off of ecosystem services. Since the implementation of the GFGP in Dali Autonomous Prefecture in 2000, land use/cover has undergone dramatic changes. This study used the CLUE-S model to simulate land use change in 2030, and explored the spatial pattern and relationship of different ecosystem services under the four scenarios of GFGP. The results show that, GFGP can help to improve indirect services of ecosystems, such as carbon storage and soil conservation. However, direct services of the ecosystem will decline, such as food production and water yield. Compared with 2010, the overall supply level of the four ecosystem services is the most balanced in the moderate GFGP scenario. In this Corresponding author E-mail address: [email protected] (J. Peng). -

Yunnan Provincial Highway Bureau
IPP740 REV World Bank-financed Yunnan Highway Assets management Project Public Disclosure Authorized Ethnic Minority Development Plan of the Yunnan Highway Assets Management Project Public Disclosure Authorized Public Disclosure Authorized Yunnan Provincial Highway Bureau July 2014 Public Disclosure Authorized EMDP of the Yunnan Highway Assets management Project Summary of the EMDP A. Introduction 1. According to the Feasibility Study Report and RF, the Project involves neither land acquisition nor house demolition, and involves temporary land occupation only. This report aims to strengthen the development of ethnic minorities in the project area, and includes mitigation and benefit enhancing measures, and funding sources. The project area involves a number of ethnic minorities, including Yi, Hani and Lisu. B. Socioeconomic profile of ethnic minorities 2. Poverty and income: The Project involves 16 cities/prefectures in Yunnan Province. In 2013, there were 6.61 million poor population in Yunnan Province, which accounting for 17.54% of total population. In 2013, the per capita net income of rural residents in Yunnan Province was 6,141 yuan. 3. Gender Heads of households are usually men, reflecting the superior status of men. Both men and women do farm work, where men usually do more physically demanding farm work, such as fertilization, cultivation, pesticide application, watering, harvesting and transport, while women usually do housework or less physically demanding farm work, such as washing clothes, cooking, taking care of old people and children, feeding livestock, and field management. In Lijiang and Dali, Bai and Naxi women also do physically demanding labor, which is related to ethnic customs. Means of production are usually purchased by men, while daily necessities usually by women. -

Teacher Shows Girls How to Reach The
18 | Friday, December 11, 2020 HONG KONG EDITION | CHINA DAILY LIFE Musical works celebrate Teacher shows girls Party-founding centenary By XING YI in Shanghai been popular since the 1970s. [email protected] Hao, a professor at the Central Conservatory of Music, says what how to reach the top Four new musical compositions he wants to express is in the poem, to mark the 100th anniversary of citing the lines: “Friends, please let the founding of the Communist us believe in the future, believe in Inspirational Party of China next year held their our unbending striving, believe in debut rehearsals in Shanghai on our youth that can conquer death, woman breaks Saturday. believe in the future — believe in The four works were created by life.” cycle of poverty musicians born in the 1950s, 60s, The last piece, My Father’s Gen- in mountain areas 70s and 80s, representing the dif- eration, was done by Yang Fan. ferent perspectives on communist Yang was the composer for the as she realizes belief from different generations. music in the award-winning dance Jia Daqun, 65, completed a con- production The Eternal Wave, potential certo for orchestra titled Zhu Lang which tells a revolutionary story of of students, Xin Chao (A Heart Pursuing the Communist intelligence’s mission Waves). The name of his music on the eve of Shanghai’s liberation Chen Meiling and echoed a 1927 poem by Mao Zed- in 1949. ong, one of the founders of the Yang says the theme of his music Li Yingqing report. CPC, which goes: I pledge my wine was drawn from the dance produc- to the surging torrent, the tide of tion, which celebrated the 70th t was a chance meeting that my heart swells with the waves. -

Phylogenetic Analysis of Organellar DNA Sequences in the Andropogoneae: Saccharinae
Theor Appl Genet (1994) 88:933-944 Springer-Verlag 1994 S. M. A1-Janabi M. McClelland C. Petersen B. W. S. Sobral Phylogenetic analysis of organellar DNA sequences in the Andropogoneae: Saccharinae Received: 30 June 1993 / Accepted: 21 December 1993 Abstract To study the phylogenetics of sugarcane (Sac- wheat, rice, barley, and maize, which were used as out- charum officinarum L.) and its relatives we sequenced four groups in phylogenetic analyses. To determine whether loci on cytoplasmic genomes (two chloroplast and two mit- limited intra-complex variability was caused by under sam- ochondrial) and analyzed mitochondrial RFLPs generated pling of taxa, we used seven restriction enzymes to digest using probes for COXI, COXII, COXIII, Cob, 18S+5S, the PCR-amplified rbcL-atpB spacer of an additional 36 26S, ATPase 6, ATPase 9, and ATPase ~ (D'Hont et al. accessions within the Saccharum complex. This analysis 1993). Approximately 650 bp of DNA in the intergenic revealed ten restriction sites (none informative) and eight spacer region between rbcL and atpB and approximately length variants (four informative). The small amount of 150 bp from the chloroplast 16S rDNA through the inter- variation present in the organellar DNAs of this polyptoid genic spacer region tRNA val gene were sequenced. In the complex suggests that either the complex is very young or mitochondrial genome, part of the 18S rRNA gene and ap- that rates of evolution between the Saccharum complex proximately 150 bp from the 18S gene 3' end, through an and outgroup taxa are different. Other phylogenetic infor- intergenic spacer region, to the 5S rRNA gene were se- mation will be required to resolve systematic relationships quenced.