Atmospheric Lifetimes of Neoteric Short-Lived Hfcs Against Oxidation
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

1,1,1,2-Tetrafluoroethane
This report contains the collective views of an international group of experts and does not necessarily represent the decisions or the stated policy of the United Nations Environment Programme, the International Labour Organisation, or the World Health Organization. Concise International Chemical Assessment Document 11 1,1,1,2-Tetrafluoroethane First draft prepared by Mrs P. Barker and Mr R. Cary, Health and Safety Executive, Liverpool, United Kingdom, and Dr S. Dobson, Institute of Terrestrial Ecology, Huntingdon, United Kingdom Please not that the layout and pagination of this pdf file are not identical to the printed CICAD Published under the joint sponsorship of the United Nations Environment Programme, the International Labour Organisation, and the World Health Organization, and produced within the framework of the Inter-Organization Programme for the Sound Management of Chemicals. World Health Organization Geneva, 1998 The International Programme on Chemical Safety (IPCS), established in 1980, is a joint venture of the United Nations Environment Programme (UNEP), the International Labour Organisation (ILO), and the World Health Organization (WHO). The overall objectives of the IPCS are to establish the scientific basis for assessment of the risk to human health and the environment from exposure to chemicals, through international peer review processes, as a prerequisite for the promotion of chemical safety, and to provide technical assistance in strengthening national capacities for the sound management of chemicals. The Inter-Organization -

Functionalization of Heterocycles: a Metal Catalyzed Approach Via
FUNCTIONALIZATION OF HETEROCYCLES: A METAL CATALYZED APPROACH VIA ALLYLATION AND C-H ACTIVATION A Dissertation Submitted to the Graduate Faculty of the North Dakota State University of Agriculture and Applied Science By Sandeepreddy Vemula In Partial Fulfillment of the Requirements for the Degree of DOCTOR OF PHILOSOPHY Major Department: Chemistry and Biochemistry October 2018 Fargo, North Dakota North Dakota State University Graduate School Title FUNCTIONALIZATION OF HETEROCYCLES: A METAL CATALYZED APPROACH VIA ALLYLATION AND C-H ACTIVATION By Sandeepreddy Vemula The Supervisory Committee certifies that this disquisition complies with North Dakota State University’s regulations and meets the accepted standards for the degree of DOCTOR OF PHILOSOPHY SUPERVISORY COMMITTEE: Prof. Gregory R. Cook Chair Prof. Mukund P. Sibi Prof. Pinjing Zhao Prof. Dean Webster Approved: November 16, 2018 Prof. Gregory R. Cook Date Department Chair ABSTRACT The central core of many biologically active natural products and pharmaceuticals contain N-heterocycles, the installation of simple/complex functional groups using C-H/N-H functionalization methodologies has the potential to dramatically increase the efficiency of synthesis with respect to resources, time and overall steps to key intermediate/products. Transition metal-catalyzed functionalization of N-heterocycles proved as a powerful tool for the construction of C-C and C-heteroatom bonds. The work in this dissertation describes the development of palladium catalyzed allylation, and the transition metal catalyzed C-H activation for selective functionalization of electron deficient N-heterocycles. Chapter 1 A thorough study highlighting the important developments made in transition metal catalyzed approaches for C-C and C-X bond forming reactions is discussed with a focus on allylation, directed indole C-2 substitution and vinylic C-H activation. -

Prepared by 3M
DRAFT ECOTOXICOLOGY AND ENVIRONMENTAL FATE TESTING OF SHORT CHAIN PERFLUOROALKYL COMPOUNDS RELATED TO 3M CHEMISTRIES XXX XX, 2008 Prepared by 3M Page 1 CONFIDENTIAL - SUBJECT TO A PROTECTIVE ORDER ENTERED IN 3M MN01537089 HENNEPIN COUNTY DISTRICT COURT, NO. 27-CV-10-28862 2231.0001 DRAFT TABLE OF CONTENTS Page Introduction (describes propose, goals, document organization .... ) 3 1) Degradation modes 4-27 Basic degradation pathways, Conversion routes from polymer to monomer & degradants or residual intermediates to degradants) 2) Degradants & Intermediates - Phys. Chem properties and test data by section & chapter i) Fluorinated Sulfonic acids and derivatives a) PFBS (PBSK) 28-42 b) PFBSI 43-44 ii) Fluorinated Carboxylates a) TFA 46-55 b) PFPA 56-59 c) PFBA (APFB) 60-65 d) MeFBSAA (MeFBSE Acid, M370) 66-68 iii) Fluorinated Non-ionics a) MeFBSE 70-74 b) MeFBSA 75-80 c) FBSE 81-84 d) FBSA 85-88 e) HxFBSA 89-91 iv) Fluorinated Inerts & volatiles a) PBSF 94-97 b) NFB, C4 hydride, CF3CF2CF2CF2-H 98-99 3) Assessments X Glossary 100-102 References/Endnotes 105- Page 2 CONFIDENTIAL - SUBJECT TO A PROTECTIVE ORDER ENTERED IN 3M MN01537090 HENNEPIN COUNTY DISTRICT COURT, NO. 27-CV-10-28862 2231.0002 DRAFT Introduction to Environmental White Papers on Perfluoroalkyl acids related to 3M chemistries Perfluorochemicals have been commonplace in chemical industry over 50 years but until recently there has been little information on environmental fate and effects avialble in open literature. The following chapters summarize the findings of"list specific C4 intermediates PFBS, PFBSI, PFBA, PFPA, MeFBSAA, TFA, MeFBSE, FBSA, HxFBSA, FBSE, PBSF, NFB " As background, 3M announced on May 16, 2000 the voluntary manufacturing phase out of perfluorooctanyl chemicals which included perfluorooctanoic acid (PFOA), perfluorooctane sulfonate (PFOS), and PFOS-related chemistries. -

Poly(Limonene Carbonate): a Bio-Based & Versatile High-Performance Thermoplastic
Poly(limonene carbonate): a bio-based & versatile high-performance thermoplastic Dissertation zur Erlangung des akademischen Grades eines Doktors der Naturwissenschaften (Dr. rer. Nat.) an der Bayreuther Graduiertenschule für Mathematik und Naturwissenschaften (BayNAT) der Universität Bayreuth vorgelegt von Oliver Hauenstein aus Dortmund Bayreuth 2016 Die vorliegende Arbeit wurde in der Zeit von November 2012 bis Juni 2016 in Bayreuth am Lehrstuhl Makromolekulare Chemie II unter Betreuung von Herrn Professor Dr. Andreas Greiner angefertigt. Vollständiger Abdruck der von der Bayreuther Graduiertenschule für Mathematik und Naturwissenschaften (BayNAT) der Universität Bayreuth genehmigten Dissertation zur Erlangung des akademischen Grades eines Doktors der Naturwissenschaften (Dr. rer. nat.). Dissertation eingereicht am: 13.07.2016 Zulassung durch das Leitungsgremium: 26.07.2016 Wissenschaftliches Kolloquium: 01.02.2017 Amtierender Direktor: Prof. Dr. Stephan Kümmel Prüfungsausschuss: Prof. Dr. Andreas Greiner (Erstgutachter) Prof. Dr. Hans-Werner Schmidt (Zweitgutachter) Prof. Dr. Carlo Unverzagt. (Vorsitz) Prof. Dr. Jürgen Senker Drittgutachter: Prof. Dr. Volker Abetz Für meine Familie & Hui Contents Abbreviations & symbols ......................................................................................................................... i List of publications ................................................................................................................................... v Abstract .................................................................................................................................................... -
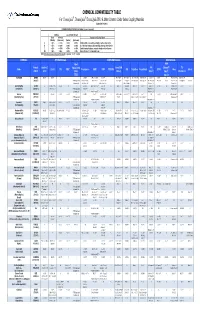
CPC Chemical Compatibility Chart
CHEMICAL COMPATIBILITY TABLE For ChemQuik®, DrumQuik®, DrumQuik PRO & Other Common Colder Series Coupling Materials (Updated 01/14/2010) INTERPRETATION OF TEST DATA (In 30 days to 1 year of exposure) Swelling Loss of Tensile Strength Linear Volumetric Description of Chemical Attack (Plastics) (Elastomers) (Plastics) (Elastomers) A < 10% <= 15% < 15% <=15% Excellent, little or no swelling, softening or surface deterioration B < 15% <= 30% < 30% <= 30% Good chemical resistance, minor swelling, softening or deterioration C < 20% <= 50% < 50% <= 60% Limited chemical resistance, moderate attack, conditional service NR > 20% > 50% > 50% > 60% Severe attack, not recommended for use NOTE: All temperatures are in degrees Fahrenheit. Conversion: °C = (°F - 32)/1.8 CHEMICAL SPRING Materials COUPLING Materials SEAL Materials Teflon® FFKM ® Formula Hastelloy C Encapsulated Acetal/POM FKM (Chemraz / TPO Name 316 SS PPS PEEK™ Polypropylene HDPE PVDF PTFE/PFA ABS Polysulfone Polycarbonate EPDM Buna Silicone (CAS #) (276) 316SS (Celcon) (Viton®) Simriz® / (Santoprene) (TESS) Kalrez®) Acetic Acid C2H4O2 A to 212° 212° A to 212° 212° A A A A to 140° 140° AB to 100% to 70° 70° A to 122° 122° A AAto5%to70 to 5% to 70°° AB 10% to 70° 70° A to 100% to 70° 70° A to 50% to 70° 70° A 10% to 70° 70° AAto70 to 70°° A B to 30% at 70° 70° A to 30% to 70° 70° A (64-19-7) (PTFE Encapsulated AB 50-100% to 160° AB 60% to 180° A to 10% to 225° BC 10% @ 70° C 20% @ 70° A to 20% to 140° B to 50% @ 122° B 10-25% to 100° AB to 200° A to 70° B to 20% to 185° C 50% @ 70° A to -

1,1,1,2-Tetrafluoroethane (HFC-134A) (CAS No. 811-97-2) (Second Edition)
1,1,1,2-Tetrafluoroethane (HFC-134a) (CAS No. 811-97-2) (Second Edition) JACC No. 50 ISSN-0773-6339-50 Brussels, January 2006 1,1,1,2-Tetrafluoroethane (HFC-134a) (CAS No. 811-97-2) (Second Edition) ECETOC JACC REPORT No. 50 © Copyright – ECETOC AISBL European Centre for Ecotoxicology and Toxicology of Chemicals 4 Avenue E. Van Nieuwenhuyse (Bte 6), B-1160 Brussels, Belgium. All rights reserved. No part of this publication may be reproduced, copied, stored in a retrieval system or transmitted in any form or by any means, electronic, mechanical, photocopying, recording or otherwise without the prior written permission of the copyright holder. Applications to reproduce, store, copy or translate should be made to the Secretary General. ECETOC welcomes such applications. Reference to the document, its title and summary may be copied or abstracted in data retrieval systems without subsequent reference. The content of this document has been prepared and reviewed by experts on behalf of ECETOC with all possible care and from the available scientific information. It is provided for information only. ECETOC cannot accept any responsibility or liability and does not provide a warranty for any use or interpretation of the material contained in the publication. ECETOC JACC No. 50 1,1,1,2-Tetrafluoroethane (HFC-134a) (CAS No. 811-97-2) (Second Edition) 1,1,1,2-Tetrafluoroethane (HFC-134a) (CAS No. 811-97-2) CONTENTS EXECUTIVE SUMMARY 1 THE ECETOC SCHEME FOR THE JOINT ASSESSMENT OF COMMODITY CHEMICALS 3 1. SUMMARY AND CONCLUSIONS 4 2. IDENTITY, PHYSICAL AND CHEMICAL PROPERTIES, ANALYTICAL METHODS 6 2.1 Identity 6 2.2 EU classification and labelling 6 2.3 Physical and chemical properties 6 2.4 Conversion factors 8 2.5 Analytical methods 8 3. -

Snider, Barry B
BARRY B. SNIDER Dept of Chemistry, MS 015 Brandeis University Waltham, MA 02454-9110 Born: 1950, Chicago Illinois Education: B. Sc. Chem. University of Michigan, Ann Arbor, MI 1970 Ph. D. Harvard University, Cambridge, MA 1973 (Advisor, E. J. Corey) Postdoctoral Columbia University, New York, NY 1973-1975 (Advisor, Ronald Breslow) Employment Assistant Professor, Princeton University, 1975-1981 Associate Professor, Brandeis University, 1981-1985 Professor, Brandeis University, 1985- Breskin Professor of Organic Chemistry, Brandeis University, 1998- Chair, Chemistry Department, Brandeis University, 1992-1995, 2013-present Awards and Honors National Science Foundation Predoctoral Fellowship 1970-1973 National Institutes of Health Postdoctoral Fellowship, 1975 DuPont Young Faculty Grant, 1978, 1979 UpJohn Young Faculty Grant, 1977, 1978 Fellow of the Alfred P. Sloan Foundation 1979-1983 Dreyfus Foundation Teacher-Scholar Award, 1982-1987 Visiting Research Scholar, Tokyo Institute of Technology, 1992 Member, NIH Medicinal Chemistry Study Section, 1992-1997 Chair, NIH Medicinal Chemistry Study Section, 1995-1997 Cope Scholar Award (American Chemical Society), 1995 Japan Society for Promotion of Science Fellowship, 1999 Honorary Professor East China University of Science and Technology 2006 Chair-Elect, Chair, Past Chair, Organic Chemistry Division of the ACS, 2006-2008 Fellow, American Chemical Society, 2011 Secretary/Treasurer, Organic Chemistry Division of the ACS, (Elect 2013) 2014-2017 Memberships: Sigma Xi, Phi Beta Kappa, American Chemical Society Link to Web page and complete publication list: Barry Snider faculty webpage Snider Publication List Page 2/24 Publications 1. Salamone, J. C.; Snider, B. “Quaternary Ammonium Polymers from 1,4- Diaza[2.2.2]bicyclooctane” J. Polym. Sci. A-1 1970, 8, 3495-3501. -

PVDF Chemical Resistance Guide
PVDF Chemical Resistance Guide SECOND EDITION PVDF CHEMICAL RESISTANCE GUIDE Thermoplastics: Kynar® Polyvinylidene Fluoride (PVDF) for Waste Drainage Systems Chemical Resistance Guide Kynar® Polyvinylidene Fluoride (PVDF) for Waste Drainage Systems 2nd Edition © 2021 by IPEX. All rights reserved. No part of this book may be used or reproduced in any manner whatsoever without prior written permission. For information contact: IPEX, Marketing, 1425 North Service Road East, Oakville, Ontario, Canada, L6H 1A7 About IPEX At IPEX, we have been manufacturing non-metallic pipe and fittings since 1951. We formulate our own compounds and maintain strict quality control during production. Our products are made available for customers thanks to a network of regional stocking locations from coast-to-coast. We offer a wide variety of systems including complete lines of piping, fittings, valves and custom-fabricated items. More importantly, we are committed to meeting our customers’ needs. As a leader in the plastic piping industry, IPEX continually develops new products, modernizes manufacturing facilities and acquires innovative process technology. In addition, our staff take pride in their work, making available to customers their extensive thermoplastic knowledge and field experience. IPEX personnel are committed to improving the safety, reliability and performance of thermoplastic materials. We are involved in several standards committees and are members of and/or comply with the organizations listed on this page. For specific details about any IPEX product, contact our customer service department. INTRODUCTION Thermoplastics and elastomers have outstanding resistance to a wide range of chemical reagents. The chemical resistance of plastic piping is basically a function of the thermoplastic material and the compounding components. -

Synthetic and Structural Studies on Natural Coumarins, THESIS
Synthetic and Structural Studies on Natural Coumarins, THESIS presented to the University of'Glasgow for the degree of Doctor of Philosophy by Rlichael Sutcliffe 1973 ProQuest Number: 11017959 All rights reserved INFORMATION TO ALL USERS The quality of this reproduction is dependent upon the quality of the copy submitted. In the unlikely event that the author did not send a com plete manuscript and there are missing pages, these will be noted. Also, if material had to be removed, a note will indicate the deletion. uest ProQuest 11017959 Published by ProQuest LLC(2018). Copyright of the Dissertation is held by the Author. All rights reserved. This work is protected against unauthorized copying under Title 17, United States C ode Microform Edition © ProQuest LLC. ProQuest LLC. 789 East Eisenhower Parkway P.O. Box 1346 Ann Arbor, Ml 48106- 1346 Acknowledgments To someone not conversant with the day-to-day atmosphere end work of an Organic Research Laboratory, the successful completion of a Ph.d thesis represents simply three years labour on behalf of its author. Nothing could be further from the truth. The very essence of successful research in Organic Chemistry lies in discussion with one’s neighbours, and also in seeking the help and advice of one’s superiors. In Glasgow, neither of these things is lacking. In particular I should like to thank my supervisor, Dr,R ,D ,H .(Hurray for his guidance, advice and unstinting help over the last three years. His enthusiasm for the subject was the source of much inspiration during my low periods. Outwith the realms of Chemistry his friendship, good humour and ready willingness to listen to any problems will be with me long after the content of this thesis is forgotten. -

Carbon–Carbon Bond Cleavage for Cu-Mediated Aromatic Trifluoromethylations and Pentafluoroethylations
Carbon–carbon bond cleavage for Cu-mediated aromatic trifluoromethylations and pentafluoroethylations Tsuyuka Sugiishi1, Hideki Amii*1, Kohsuke Aikawa2 and Koichi Mikami*2 Review Open Access Address: Beilstein J. Org. Chem. 2015, 11, 2661–2670. 1Division of Molecular Science, Faculty of Science and Technology, doi:10.3762/bjoc.11.286 Gunma University, 1-5-1 Tenjin-cho, Kiryu, Gunma 376-8515, Japan and 2Department of Applied Chemistry, Graduate School of Science Received: 30 September 2015 and Engineering, Tokyo Institute of Technology, O-okayama, Accepted: 11 December 2015 Meguro-ku, Tokyo 152-8552, Japan Published: 18 December 2015 Email: This article is part of the Thematic Series "Copper catalysis in organic Hideki Amii* - [email protected]; Koichi Mikami* - synthesis". [email protected] Guest Editor: S. R. Chemler * Corresponding author © 2015 Sugiishi et al; licensee Beilstein-Institut. Keywords: License and terms: see end of document. β-carbon elimination; carbon–carbon bond cleavage; decarboxylation; tetrahedral intermediate; trifluoroacetate; fluoral; trifluoromethylation Abstract This short review highlights the copper-mediated fluoroalkylation using perfluoroalkylated carboxylic acid derivatives. Carbon–carbon bond cleavage of perfluoroalkylated carboxylic acid derivatives takes place in fluoroalkylation reactions at high temperature (150–200 °C) or under basic conditions to generate fluoroalkyl anion sources for the formation of fluoroalkylcopper species. The fluoroalkylation reactions, which proceed through decarboxylation or tetrahedral intermediates, are useful protocols for the synthesis of fluoroalkylated aromatics. Introduction Organofluorine compounds attract attention because of their ap- biologically active CF3-containing molecules [8]. Perfluoro- plicability in various fields, such as medicine, agrochemical and alkylcopper compounds (CnF2n+1Cu), which are soft and rela- material science. -
—*W^U Formate with a Drop of Trifluoroacetic Acid Was Re- XXX Fluxed for 0.5 to 3 H
A Simple Route to /V-Methylarylamines Z. DASZKIEWICZ, A. DOMAŇSKI, and J. B. KYZIOt Instytut Chemii, Wyžsza Szkoia Pedagogiczna, PL-45 052 Opole Received 9 April 1992 Thirteen ring-substituted anilines were transformed into corresponding formanilides by the action of n-butyl formate in the presence of trifluoroacetic acid and then reduced with lithium aluminium hydride to /V-methylanilines in total yields 40—70 %. The method can be employed for methylation of primary and secondary amino groups bound to aromatic and heteroaromatic systems provided that they are neither sterically hindered nor strongly conjugated with other substituents. The general method of /V-alkylamines preparation, EXPERIMENTAL i.e. alkylation with alkyl halides or inorganic acids esters and reductive alkylations with carbonyl com The starting, amines were commercial reagents, pounds are of limited value as regards synthesis of pure, used without purification. The progress of re /V-methylaniline and its derivatives. Reaction of ani actions was followed by the TLC method (Silufol; line with methyl iodide affords the quaternary am Kavalier, Votice) using PhMe—AcOEt (<pr = 2 : 1) and monium salt [1], with less reactive methyl sulfate CHCI3—MeOH («ft = 24 : 1 to 5 : 1) mixtures as the a mixture of compounds is obtained from which developing systems; the plates were sprayed with /V-methylaniline can be isolated as the /V-nitroso de 1 % rhodamine В and observed under the UV lamp. rivative [2]. Methylation of /V-tosylaniline presents no All the compounds gave satisfactory elemental such problems but the cleavage of the sulfonamide analyses. The purity of /V-methylarylamines was bond requires very severe conditions [3], hence only controlled on the gas Chromatograph 900 some aniline derivatives can be /V-methylated in this (Perkin—Elmer) equipped with the flame ionization way. -

Isolation and Functional Characterization of CYP71AJ4
Isolation and Functional Characterization of CYP71AJ4 Encoding for the First P450 Monooxygenase of Angular Furanocoumarin Biosynthesis Romain Larbat, Alain Hehn, Joachim Hans, Sarah Schneider, Hélène Jugdé, Bernd Schneider, Ulrich Matern, Frédéric Bourgaud To cite this version: Romain Larbat, Alain Hehn, Joachim Hans, Sarah Schneider, Hélène Jugdé, et al.. Isolation and Functional Characterization of CYP71AJ4 Encoding for the First P450 Monooxygenase of Angular Furanocoumarin Biosynthesis. Journal of Biological Chemistry, American Society for Biochemistry and Molecular Biology, 2009, 284 (8), pp.4776-4785. 10.1074/jbc.M807351200. hal-01738063 HAL Id: hal-01738063 https://hal.archives-ouvertes.fr/hal-01738063 Submitted on 20 Mar 2018 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. THE JOURNAL OF BIOLOGICAL CHEMISTRY VOL. 284, NO. 8, pp. 4776–4785, February 20, 2009 © 2009 by The American Society for Biochemistry and Molecular Biology, Inc. Printed in the U.S.A. Isolation and Functional Characterization of CYP71AJ4 Encoding for the First P450 Monooxygenase