The Role of 1,3-Dithianes in Natural Product Synthesis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

1,1,1,2-Tetrafluoroethane
This report contains the collective views of an international group of experts and does not necessarily represent the decisions or the stated policy of the United Nations Environment Programme, the International Labour Organisation, or the World Health Organization. Concise International Chemical Assessment Document 11 1,1,1,2-Tetrafluoroethane First draft prepared by Mrs P. Barker and Mr R. Cary, Health and Safety Executive, Liverpool, United Kingdom, and Dr S. Dobson, Institute of Terrestrial Ecology, Huntingdon, United Kingdom Please not that the layout and pagination of this pdf file are not identical to the printed CICAD Published under the joint sponsorship of the United Nations Environment Programme, the International Labour Organisation, and the World Health Organization, and produced within the framework of the Inter-Organization Programme for the Sound Management of Chemicals. World Health Organization Geneva, 1998 The International Programme on Chemical Safety (IPCS), established in 1980, is a joint venture of the United Nations Environment Programme (UNEP), the International Labour Organisation (ILO), and the World Health Organization (WHO). The overall objectives of the IPCS are to establish the scientific basis for assessment of the risk to human health and the environment from exposure to chemicals, through international peer review processes, as a prerequisite for the promotion of chemical safety, and to provide technical assistance in strengthening national capacities for the sound management of chemicals. The Inter-Organization -

Functionalization of Heterocycles: a Metal Catalyzed Approach Via
FUNCTIONALIZATION OF HETEROCYCLES: A METAL CATALYZED APPROACH VIA ALLYLATION AND C-H ACTIVATION A Dissertation Submitted to the Graduate Faculty of the North Dakota State University of Agriculture and Applied Science By Sandeepreddy Vemula In Partial Fulfillment of the Requirements for the Degree of DOCTOR OF PHILOSOPHY Major Department: Chemistry and Biochemistry October 2018 Fargo, North Dakota North Dakota State University Graduate School Title FUNCTIONALIZATION OF HETEROCYCLES: A METAL CATALYZED APPROACH VIA ALLYLATION AND C-H ACTIVATION By Sandeepreddy Vemula The Supervisory Committee certifies that this disquisition complies with North Dakota State University’s regulations and meets the accepted standards for the degree of DOCTOR OF PHILOSOPHY SUPERVISORY COMMITTEE: Prof. Gregory R. Cook Chair Prof. Mukund P. Sibi Prof. Pinjing Zhao Prof. Dean Webster Approved: November 16, 2018 Prof. Gregory R. Cook Date Department Chair ABSTRACT The central core of many biologically active natural products and pharmaceuticals contain N-heterocycles, the installation of simple/complex functional groups using C-H/N-H functionalization methodologies has the potential to dramatically increase the efficiency of synthesis with respect to resources, time and overall steps to key intermediate/products. Transition metal-catalyzed functionalization of N-heterocycles proved as a powerful tool for the construction of C-C and C-heteroatom bonds. The work in this dissertation describes the development of palladium catalyzed allylation, and the transition metal catalyzed C-H activation for selective functionalization of electron deficient N-heterocycles. Chapter 1 A thorough study highlighting the important developments made in transition metal catalyzed approaches for C-C and C-X bond forming reactions is discussed with a focus on allylation, directed indole C-2 substitution and vinylic C-H activation. -

Carbonyl Compounds
CARBONYL COMPOUNDS PART-4, PPT-4, SEM-3 Dr. Kalyan Kumar Mandal Associate Professor St. Paul’s C. M. College Kolkata CONTENTS: CARBONYL COMPOUNDS PART-4 • Formation of Acetal/Ketal • Formation of Thioacetal Reaction of Carbonyl Compounds with Alcohols • Carbonyl compounds react with alcohols. The product of this reaction is known as a hemiacetal, because it is halfway to an acetal. This reaction is analogous to hydrate formation from aldehydes and ketones. The mechanism follows in the footsteps of hydrate formation: ROH is used instead of HOH (water). This Lecture is prepared by Dr. K. K. Mandal, SPCMC, Kolkata Formation of Cyclic Hemiacetal • Hemiacetal formation is reversible, and they are stabilized by the same special structural features as those of hydrates. However, hemiacetals can also gain stability by being cyclic. • Cyclic hemiacetal is formed when the carbonyl group and the attacking hydroxyl group are part of the same molecule. The reaction is an intramolecular (within the same molecule) addition, as opposed to the intermolecular (between two molecules) ones. This is an example of ring-chain tautomerism. This Lecture is prepared by Dr. K. K. Mandal, SPCMC, Kolkata Formation of Cyclic Hemiacetal • Although the cyclic hemiacetal is more stable, it is still in equilibrium with some of the open-chain hydroxyaldehyde form. Its stability, and how easily it forms, depend on the size of the ring. • Five- and six-membered rings involve less strain (their bonds are free to adopt 109° or 120° angles) in comparison to the three- membered rings, and therefore five or six-membered hemiacetals are very common. -

TABLE 18-1 Some Common Classes of Carbonyl Compounds
TABLE 18-1 Some Common Classes of Carbonyl Compounds Class General Formula Class General Formula O O ' ' ketones R9C9RЈ aldehydes R9C9H O O ' ' carboxylic acids R9C9OH acid chlorides R9C9Cl O O ' ' 9 9 9 Ј 9 9 esters R C O R amides R C NH2 AAALBKS0 Figure Number: 18 00.01 T01 ©2003 by Prentice Hall, Inc. WADE A Pearson Company ORGANIC CHEMISTRY, 5E length energy ketone CObond 1.23 Å 178 kcal/mol R (745 kJ/mol) 120° C O R 120° alkene C C bond 1.34 Å 146 kcal/mol (611 kJ/mol) AAALBKU0 Figure Number: 18 00.03 UN ©2003 by Prentice Hall, Inc. WADE A Pearson Company ORGANIC CHEMISTRY, 5E R R Ϫ C O ϩC O R R major minor AAALBKV0 Figure Number: 18 00.04 UN ©2003 by Prentice Hall, Inc. WADE A Pearson Company ORGANIC CHEMISTRY, 5E O O 9 9 9 9 9 9 9 CH3CH2CH2CH3 CH3 O CH2CH3 CH3CH2 C H CH3 C CH3 CH3CH2CH2 OH butane methoxyethane propanal acetone 1-propanol bp 0°C bp 8°C bp 49°C bp 56°C bp 97°C AAALBLI0 Figure Number: 18 00.17 UN 1-5 ©2003 by Prentice Hall, Inc. WADE A Pearson Company ORGANIC CHEMISTRY, 5E about 1685 cmϪ1 O OϪ C C C C ϩ C C O 1685 cmϪ1 O 1690 cmϪ1 O 1745 cmϪ1 O 1815 cmϪ1 C CH3 H acetophenone 2-butenal cyclopentanone cyclopropanone AAALBLM0 Figure Number: 18 00.22 UN ©2003 by Prentice Hall, Inc. WADE A Pearson Company ORGANIC CHEMISTRY, 5E a carbon a carbon a carbon O O O Ј R CH2 C H R C CH3 R C CH2R dd2.4 9–d10 d2.1 d2.4 an aldehyde a methyl ketone other ketones AAALBLN0 Figure Number: 18 00.23 UN 1-3 ©2003 by Prentice Hall, Inc. -

Prepared by 3M
DRAFT ECOTOXICOLOGY AND ENVIRONMENTAL FATE TESTING OF SHORT CHAIN PERFLUOROALKYL COMPOUNDS RELATED TO 3M CHEMISTRIES XXX XX, 2008 Prepared by 3M Page 1 CONFIDENTIAL - SUBJECT TO A PROTECTIVE ORDER ENTERED IN 3M MN01537089 HENNEPIN COUNTY DISTRICT COURT, NO. 27-CV-10-28862 2231.0001 DRAFT TABLE OF CONTENTS Page Introduction (describes propose, goals, document organization .... ) 3 1) Degradation modes 4-27 Basic degradation pathways, Conversion routes from polymer to monomer & degradants or residual intermediates to degradants) 2) Degradants & Intermediates - Phys. Chem properties and test data by section & chapter i) Fluorinated Sulfonic acids and derivatives a) PFBS (PBSK) 28-42 b) PFBSI 43-44 ii) Fluorinated Carboxylates a) TFA 46-55 b) PFPA 56-59 c) PFBA (APFB) 60-65 d) MeFBSAA (MeFBSE Acid, M370) 66-68 iii) Fluorinated Non-ionics a) MeFBSE 70-74 b) MeFBSA 75-80 c) FBSE 81-84 d) FBSA 85-88 e) HxFBSA 89-91 iv) Fluorinated Inerts & volatiles a) PBSF 94-97 b) NFB, C4 hydride, CF3CF2CF2CF2-H 98-99 3) Assessments X Glossary 100-102 References/Endnotes 105- Page 2 CONFIDENTIAL - SUBJECT TO A PROTECTIVE ORDER ENTERED IN 3M MN01537090 HENNEPIN COUNTY DISTRICT COURT, NO. 27-CV-10-28862 2231.0002 DRAFT Introduction to Environmental White Papers on Perfluoroalkyl acids related to 3M chemistries Perfluorochemicals have been commonplace in chemical industry over 50 years but until recently there has been little information on environmental fate and effects avialble in open literature. The following chapters summarize the findings of"list specific C4 intermediates PFBS, PFBSI, PFBA, PFPA, MeFBSAA, TFA, MeFBSE, FBSA, HxFBSA, FBSE, PBSF, NFB " As background, 3M announced on May 16, 2000 the voluntary manufacturing phase out of perfluorooctanyl chemicals which included perfluorooctanoic acid (PFOA), perfluorooctane sulfonate (PFOS), and PFOS-related chemistries. -

Enantioselective Organocatalytic Aldehyde–Aldehyde Cross-Aldol Couplings
Tetrahedron 60 (2004) 7705–7714 Enantioselective organocatalytic aldehyde–aldehyde cross-aldol couplings. The broad utility of a-thioacetal aldehydes R. Ian Storer and David W. C. MacMillan* Division of Chemistry and Chemical Engineering, California Institute of Technology, 1200 E California Blvd; Pasadena, CA 91125, USA Received 6 April 2004; accepted 9 April 2004 Available online 15 July 2004 This manuscript is dedicated to Professor D. Seebach for his pioneering work in the area of asymmetric synthesis Abstract—An asymmetric proline catalyzed aldol reaction with a-thioacetal aldehydes has been developed. Thioacetal bearing aldehydes readily participate as electrophilic cross-aldol partners with a broad range of aldehyde and ketone donors. High levels of reaction efficiency as well as diastereo- and enantiocontrol are observed in the production of anti-aldol adducts. q 2004 Elsevier Ltd. All rights reserved. 1. Introduction enantioselective cross coupling allows access to highly oxidized, stereodefined synthons of broad versatility. More- The aldol reaction is widely considered to be one of the most over, the observed reactivity profile of ‘viable-electrophile, important technologies for carbon–carbon bond formation non-nucleophile’ has established a-thioacetal aldehydes as in chemical synthesis.1 Over the last thirty years, seminal research from the laboratories of Evans,2 Heathcock,3 Masamune4 and Mukaiyama5 have established this vener- able reaction as the principal chemical method for the stereoselective construction of complex polyol architecture. Recently, studies by Barbas,6 Evans,7 List,8 Shair,9 Shibasaki,10 and Trost11 have outlined the first examples of enantioselective direct aldol reactions, an important class of metal or proline catalyzed transformation that does not require the pregeneration of enolates or enolate equivalents. -

Poly(Limonene Carbonate): a Bio-Based & Versatile High-Performance Thermoplastic
Poly(limonene carbonate): a bio-based & versatile high-performance thermoplastic Dissertation zur Erlangung des akademischen Grades eines Doktors der Naturwissenschaften (Dr. rer. Nat.) an der Bayreuther Graduiertenschule für Mathematik und Naturwissenschaften (BayNAT) der Universität Bayreuth vorgelegt von Oliver Hauenstein aus Dortmund Bayreuth 2016 Die vorliegende Arbeit wurde in der Zeit von November 2012 bis Juni 2016 in Bayreuth am Lehrstuhl Makromolekulare Chemie II unter Betreuung von Herrn Professor Dr. Andreas Greiner angefertigt. Vollständiger Abdruck der von der Bayreuther Graduiertenschule für Mathematik und Naturwissenschaften (BayNAT) der Universität Bayreuth genehmigten Dissertation zur Erlangung des akademischen Grades eines Doktors der Naturwissenschaften (Dr. rer. nat.). Dissertation eingereicht am: 13.07.2016 Zulassung durch das Leitungsgremium: 26.07.2016 Wissenschaftliches Kolloquium: 01.02.2017 Amtierender Direktor: Prof. Dr. Stephan Kümmel Prüfungsausschuss: Prof. Dr. Andreas Greiner (Erstgutachter) Prof. Dr. Hans-Werner Schmidt (Zweitgutachter) Prof. Dr. Carlo Unverzagt. (Vorsitz) Prof. Dr. Jürgen Senker Drittgutachter: Prof. Dr. Volker Abetz Für meine Familie & Hui Contents Abbreviations & symbols ......................................................................................................................... i List of publications ................................................................................................................................... v Abstract .................................................................................................................................................... -
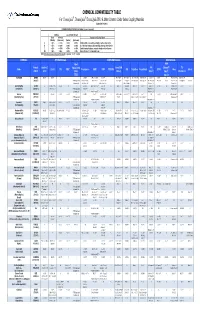
CPC Chemical Compatibility Chart
CHEMICAL COMPATIBILITY TABLE For ChemQuik®, DrumQuik®, DrumQuik PRO & Other Common Colder Series Coupling Materials (Updated 01/14/2010) INTERPRETATION OF TEST DATA (In 30 days to 1 year of exposure) Swelling Loss of Tensile Strength Linear Volumetric Description of Chemical Attack (Plastics) (Elastomers) (Plastics) (Elastomers) A < 10% <= 15% < 15% <=15% Excellent, little or no swelling, softening or surface deterioration B < 15% <= 30% < 30% <= 30% Good chemical resistance, minor swelling, softening or deterioration C < 20% <= 50% < 50% <= 60% Limited chemical resistance, moderate attack, conditional service NR > 20% > 50% > 50% > 60% Severe attack, not recommended for use NOTE: All temperatures are in degrees Fahrenheit. Conversion: °C = (°F - 32)/1.8 CHEMICAL SPRING Materials COUPLING Materials SEAL Materials Teflon® FFKM ® Formula Hastelloy C Encapsulated Acetal/POM FKM (Chemraz / TPO Name 316 SS PPS PEEK™ Polypropylene HDPE PVDF PTFE/PFA ABS Polysulfone Polycarbonate EPDM Buna Silicone (CAS #) (276) 316SS (Celcon) (Viton®) Simriz® / (Santoprene) (TESS) Kalrez®) Acetic Acid C2H4O2 A to 212° 212° A to 212° 212° A A A A to 140° 140° AB to 100% to 70° 70° A to 122° 122° A AAto5%to70 to 5% to 70°° AB 10% to 70° 70° A to 100% to 70° 70° A to 50% to 70° 70° A 10% to 70° 70° AAto70 to 70°° A B to 30% at 70° 70° A to 30% to 70° 70° A (64-19-7) (PTFE Encapsulated AB 50-100% to 160° AB 60% to 180° A to 10% to 225° BC 10% @ 70° C 20% @ 70° A to 20% to 140° B to 50% @ 122° B 10-25% to 100° AB to 200° A to 70° B to 20% to 185° C 50% @ 70° A to -

Publikationsliste Prof. Dr. D. Seebach
Publikationsliste Prof. Dr. D. Seebach 1 + Diplomarbeit: Dieter Seebach Zur Reaktion von Bleitetraacetat mit 1,1-Diphenyl-2-hydroperoxy-propiomesitylen Technische Hochschule Karlsruhe, 1961 2 + Dissertation: Dieter Seebach 2.5-Dihydro-Furan-Peroxyde Technische Hochschule Karlsruhe, 1964 3 Rudolf Criegee, Dieter Seebach Ein Bishydroperoxyd mit ungewöhlicher Bildungstendenz Chem. Ber. 96, 2704 - 2711 (1963) 4 Dieter Seebach Die Reaktion von 2.5-Dimethyl-furan mit Wasserstoffperoxyd Chem. Ber. 96, 2712 - 2722 (1963) 5 Dieter Seebach Die Reaktion von Pentamethylpyrrol mit Wasserstoffperoxyd Chem. Ber. 96, 2723 - 2729 (1963) 6 Rudolf Criegee, Ulrich Zirngibl, Harald Furrer, Dieter Seebach, Günther Freund Photosynthese substituierter Cyclobutene Chem. Ber. 97, 2942 - 2948 (1964) 7 Dieter Seebach Über ein sehr labiles Bicyclo(2.2.0)hexen-Derivat Chem. Ber. 97, 2953 - 2958 (1964) 8 * Dieter Seebach Gespannte polycyclische Systeme aus Drei-und Vierring-Bausteinen Angew. Chem. 77, 119 - 129 (1965) Angew. Chem. Int. Ed. Engl. 4, 121 - 131 (1965) 9 Rudolf Criegee, Haukur Kristinsson, Dieter Seebach, Fritz Zanker Eine neuartige Synthese von Bicyclo(2.2.0)hexen-(2)-Derivaten Chem. Ber. 98, 2331 - 2338 (1965) 10 Rudolf Criegee, Dieter Seebach, Rudolf Ernst Winter, Bernt Börretzen, Hans-Albert Brune Valenzisomerisierungen von Cyclobutenen Chem. Ber. 98, 2339 - 2352 (1965) 11 Elias J. Corey, Dieter Seebach Carbanionen der 1,3-Dithiane, Reagentien zur C-C-Verknüpfung durch nucleophile Substitution oder Carbonyl-Addition Angew. Chem. 77, 1134 - 1135 (1965) Angew. Chem. Int. Ed. Engl. 4, 1075 - 1077 (1965) 12 Elias J. Corey, Dieter Seebach Synthese von 1,n-Dicarbonylverbindungen mit Carbanionen der 1,3-Dithiane Angew. Chem. 77, 1135 - 1136 (1965) Angew. -
![[Beta]-Keto Sulfoxides Leo Arthur Ochrymowycz Iowa State University](https://docslib.b-cdn.net/cover/9355/beta-keto-sulfoxides-leo-arthur-ochrymowycz-iowa-state-university-2519355.webp)
[Beta]-Keto Sulfoxides Leo Arthur Ochrymowycz Iowa State University
Iowa State University Capstones, Theses and Retrospective Theses and Dissertations Dissertations 1969 Chemistry of [beta]-keto sulfoxides Leo Arthur Ochrymowycz Iowa State University Follow this and additional works at: https://lib.dr.iastate.edu/rtd Part of the Organic Chemistry Commons Recommended Citation Ochrymowycz, Leo Arthur, "Chemistry of [beta]-keto sulfoxides " (1969). Retrospective Theses and Dissertations. 3766. https://lib.dr.iastate.edu/rtd/3766 This Dissertation is brought to you for free and open access by the Iowa State University Capstones, Theses and Dissertations at Iowa State University Digital Repository. It has been accepted for inclusion in Retrospective Theses and Dissertations by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. This dissertation has been microfihned exactly as received 70-7726 OCHRYMOWYCZ, Leo Arthur, 1943- CHEMISTRY OF p -KETO SULFOXIDES. Iowa State University, Ph.D., 1969 Chemistry, organic University Microfilms, Inc., Ann Arbor, Michigan CHEMISTRY OF jg-KETO SULFOXIDES by Leo Arthur Ochrymowycz A Dissertation Submitted to the Graduate Faculty in Partial Fulfillment of The Requirements for the Degree of DOCTOR OF PHILOSOPHY Major Subject ; Organic Chemistry Approved : Signature was redacted for privacy. f Major Work Signature was redacted for privacy. d of Maj Department Signature was redacted for privacy. Graduate College Iowa State University Of Science and Technology Ames, Iowa 1969 il TABLE OP CONTENTS Page -

1,1,1,2-Tetrafluoroethane (HFC-134A) (CAS No. 811-97-2) (Second Edition)
1,1,1,2-Tetrafluoroethane (HFC-134a) (CAS No. 811-97-2) (Second Edition) JACC No. 50 ISSN-0773-6339-50 Brussels, January 2006 1,1,1,2-Tetrafluoroethane (HFC-134a) (CAS No. 811-97-2) (Second Edition) ECETOC JACC REPORT No. 50 © Copyright – ECETOC AISBL European Centre for Ecotoxicology and Toxicology of Chemicals 4 Avenue E. Van Nieuwenhuyse (Bte 6), B-1160 Brussels, Belgium. All rights reserved. No part of this publication may be reproduced, copied, stored in a retrieval system or transmitted in any form or by any means, electronic, mechanical, photocopying, recording or otherwise without the prior written permission of the copyright holder. Applications to reproduce, store, copy or translate should be made to the Secretary General. ECETOC welcomes such applications. Reference to the document, its title and summary may be copied or abstracted in data retrieval systems without subsequent reference. The content of this document has been prepared and reviewed by experts on behalf of ECETOC with all possible care and from the available scientific information. It is provided for information only. ECETOC cannot accept any responsibility or liability and does not provide a warranty for any use or interpretation of the material contained in the publication. ECETOC JACC No. 50 1,1,1,2-Tetrafluoroethane (HFC-134a) (CAS No. 811-97-2) (Second Edition) 1,1,1,2-Tetrafluoroethane (HFC-134a) (CAS No. 811-97-2) CONTENTS EXECUTIVE SUMMARY 1 THE ECETOC SCHEME FOR THE JOINT ASSESSMENT OF COMMODITY CHEMICALS 3 1. SUMMARY AND CONCLUSIONS 4 2. IDENTITY, PHYSICAL AND CHEMICAL PROPERTIES, ANALYTICAL METHODS 6 2.1 Identity 6 2.2 EU classification and labelling 6 2.3 Physical and chemical properties 6 2.4 Conversion factors 8 2.5 Analytical methods 8 3. -

Sulfur-Facilitated Organic Synthesis
Sulfur-Facilitated Organic Synthesis Andrew McClory Monday March 16, 2009 8:00 pm, 147 Noyes S S S S S S 'Attempts to make thioacetone by the cracking of trithioacetone gave rise to an offensive smell which spread rapidly over a great area of the town causing fainting, vomiting and a panic evacuation'...'the laboratory work was abandoned.' -Researcher, Freiburg, 1889 S S S 'Attempts to make thioacetone by the cracking of trithioacetone gave rise to an offensive smell which spread rapidly over a great area of the town causing fainting, vomiting and a panic evacuation'...'the laboratory work was abandoned.' -Researcher, Freiburg, 1889 'Recently we found ourselves with an odour problem beyond our worst expectations. During early experiments, a stopper jumped from a bottle of residues, and, although replaced at once, resulted in an immediate complaint of nausea and sickness from colleagues working in a building two hundred yards away. Two of our chemists who had done no more than investigate the cracking of minute amounts of trithioacetone found themselves the object of hostile stares in a restaurant and suffered the humiliation of having a waitress spray the area around them with a deodorant. The odours defied the expected effects of dilution since workers in the laboratory did not find the odours intolerable...and genuinely denied responsibility since they were working in closed systems. To convince them otherwise, they were dispersed with other observers around the laboratory, at distances up to a quarter of a mile, and one drop of either acetone gem-dithiol or the mother liquors from crude thioacetone crystallizations were placed on a watch glass in a fume cupboard.