Polypivalolactone
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Alembic Pot Still
ALEMBIC POT STILL INSTRUCTION MANUAL CAN BE USED WITH THE GRAINFATHER OR T500 BOILER SAFETY Warning: This system produces a highly flammable liquid. PRECAUTION: • Always use the Alembic Pot Still System in a room with adequate ventilation. • Never leave the Alembic Pot Still system unattended when operating. • Keep the Alembic Pot Still system away from all sources of ignition, including smoking, sparks, heat, and open flames. • Ensure all other equipment near to the Alembic Pot Still system or the alcohol is earthed. • A fire extinguishing media suitable for alcohol should be kept nearby. This can be water fog, fine water spray, foam, dry powder, carbon dioxide, sand or dolomite. • Do not boil dry. In the event the still is boiled dry, reset the cutout button under the base of the still. In the very unlikely event this cutout fails, a fusible link gives an added protection. IN CASE OF SPILLAGE: • Shut off all possible sources of ignition. • Clean up spills immediately using cloth, paper towels or other absorbent materials such as soil, sand or other inert material. • Collect, seal and dispose accordingly • Mop area with excess water. CONTENTS Important points before getting started ............................................................................... 3 Preparing the Alembic Pot Still ................................................................................................. 5 Distilling a Whiskey, Rum or Brandy .......................................................................................7 Distilling neutral -

Minimum Reflux in Liquid–Liquid Extraction
17th European Symposium on Computer Aided Process Engineering – ESCAPE17 V. Plesu and P.S. Agachi (Editors) © 2007 Elsevier B.V. All rights reserved. 1 Minimum Reflux in Liquid–Liquid Extraction Santanu Bandyopadhyaya and Calin-Cristian Cormosb aEnergy Systems Engineering, Indian Institute of Technology, Bombay, Powai, Mumbai 400 076, India, E-mail: [email protected] bDepartment of Chemical Engineering, Faculty of Chemistry and Chemical Engineering, University "Babeş-Bolyai", Arany Janos 11, Cluj-Napoca 400028, Romania, E-mail: [email protected] Abstract In a simple countercurrent arrangement of different stages of liquid–liquid extraction operation, the richest extract leaving the operation is in equilibrium with the feed. However, by using reflux it is possible to enrich the extract further. A simple counter current liquid–liquid extraction operation with reflux is analogous in its essentials to distillation. In this paper, the method based on Invariant Rectifying-Stripping (IRS) curves, originally proposed to calculate minimum reflux and minimum energy requirement in distillation, has been extended to liquid–liquid extraction. The equivalent IRS curves for a ternary liquid–liquid extraction predicts the feed location in the counter current process. It also predicts the minimum reflux requirement for a given separation and minimum amount of solvent required. Keywords: liquid extraction, IRS curves, feed location, minimum solvent. 1. Introduction Liquid–liquid extraction is a separation process that takes advantage of the distribution of a substance between two insoluble liquids phases [1]. Feed to be separated is mixed with extracting solvent to produce a solvent-rich phase, called extract, and a solvent-lean phase, called raffinate. -

Reflux Condensation in Narrow Rectangular Channels with Perforated Fins Nadia Souidi, André Bontemps
Reflux condensation in narrow rectangular channels with perforated fins Nadia Souidi, André Bontemps To cite this version: Nadia Souidi, André Bontemps. Reflux condensation in narrow rectangular channels with perforated fins. Applied Thermal Engineering, Elsevier, 2003, 23, pp.871-891. 10.1016/S1359-4311(03)00021-8. hal-00184135 HAL Id: hal-00184135 https://hal.archives-ouvertes.fr/hal-00184135 Submitted on 19 Feb 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Reflux condensation in narrow rectangular channels with perforated fins N. Souidi a, A. Bontemps b,* a GRETh-CEA Grenoble, 17 Rue des Martyrs, 38054 Grenoble Cedex 9, France b LEGI-GRETh, Universitee Joseph Fourier, 17 Rue des Martyrs, 38054 Grenoble Cedex 9, France Reflux condensation is an industrial process that aims to reduce the content of the less volatile com- ponent or to eliminate the non-condensable phase of a vapour mixture, by the means of separation. Separation consists in condensing the less volatile phase and to recover the condensate while simulta- neously, the non-condensable species are recuperated at the top of the system. Compact plate-fin heat exchangers can be used in gas separation processes. -

Building a Home Distillation Apparatus
BUILDING A HOME DISTILLATION APPARATUS A Step by Step Guide Building a Home Distillation Apparatus i BUILDING A HOME DISTILLATION APPARATUS Foreword The pages that follow contain a step-by-step guide to building a relatively sophisticated distillation apparatus from commonly available materials, using simple tools, and at a cost of under $100 USD. The information contained on this site is directed at anyone who may want to know more about the subject: students, hobbyists, tinkers, pure water enthusiasts, survivors, the curious, and perhaps even amateur wine and beer makers. Designing and building this apparatus is the only subject of this manual. You will find that it confines itself solely to those areas. It does not enter into the domains of fermentation, recipes for making mash, beer, wine or any other spirits. These areas are covered in detail in other readily available books and numerous web sites. The site contains two separate design plans for the stills. And while both can be used for a number of distillation tasks, it should be recognized that their designs have been optimized for the task of separating ethyl alcohol from a water-based mixture. Having said that, remember that the real purpose of this site is to educate and inform those of you who are interested in this subject. It is not to be construed in any fashion as an encouragement to break the law. If you believe the law is incorrect, please take the time to contact your representatives in government, cast your vote at the polls, write newsletters to the media, and in general, try to make the changes in a legal and democratic manner. -

Batch Distillation of Spirits: Experimental Study and Simulation
Research article Received: 5 April 2018 Revised: 11 January 2019 Accepted: 15 January 2019 Published online in Wiley Online Library (wileyonlinelibrary.com) DOI 10.1002/jib.560 Batch distillation of spirits: experimental study and simulation of the behaviour of volatile aroma compounds Adrien Douady,1 Cristian Puentes,1 Pierre Awad1,2 and Martine Esteban-Decloux1* This paper focuses on the behaviour of volatile compounds during batch distillation of wine or low wine, in traditional Charentais copper stills, heated with a direct open flame at laboratory (600 L) and industrial (2500 L) scale. Sixty-nine volatile compounds plus ethanol were analysed during the low wine distillation in the 600 L alembic still. Forty-four were quantified and classified according to their concentration profile in the distillate over time and compared with previous studies. Based on the online re- cording of volume flow, density and temperature of the distillate with a Coriolis flowmeter, distillation was simulated with ProSim® BatchColumn software. Twenty-six volatile compounds were taken into account, using the coefficients of the ‘Non- Random Two Liquids’ model. The concentration profiles of 18 compounds were accurately represented, with slight differences in the maximum concentration for seven species together with a single compound that was poorly represented. The distribution of the volatile compounds in the four distillate fractions (heads, heart, seconds and tails) was well estimated by simulation. Fi- nally, data from wine and low wine distillations in the large-scale alembic still (2500 L) were correctly simulated, suggesting that it was possible to adjust the simulation parameters with the Coriolis flowmeter recording and represent the concentration pro- files of most of the quantifiable volatile compounds. -
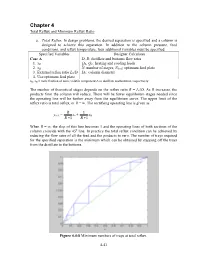
Minimum Reflux Ratio
Chapter 4 Total Reflux and Minimum Reflux Ratio a. Total Reflux . In design problems, the desired separation is specified and a column is designed to achieve this separation. In addition to the column pressure, feed conditions, and reflux temperature, four additional variables must be specified. Specified Variables Designer Calculates Case A D, B: distillate and bottoms flow rates 1. xD QR, QC: heating and cooling loads 2. xB N: number of stages, Nfeed : optimum feed plate 3. External reflux ratio L0/D DC: column diameter 4. Use optimum feed plate xD, xB = mole fraction of more volatile component A in distillate and bottoms, respectively The number of theoretical stages depends on the reflux ratio R = L0/D. As R increases, the products from the column will reduce. There will be fewer equilibrium stages needed since the operating line will be further away from the equilibrium curve. The upper limit of the reflux ratio is total reflux, or R = ∞. The rectifying operating line is given as R 1 yn+1 = xn + xD R +1 R +1 When R = ∞, the slop of this line becomes 1 and the operating lines of both sections of the column coincide with the 45 o line. In practice the total reflux condition can be achieved by reducing the flow rates of all the feed and the products to zero. The number of trays required for the specified separation is the minimum which can be obtained by stepping off the trays from the distillate to the bottoms. Figure 4.4-8 Minimum numbers of trays at total reflux. -

Members/Downloads/Docs/Distilling Instructions Super Reflux Condenser.Pdf
Distilling Instructions (Super Reflux Condenser) 9 Adams Road Palmerston NT 0830 Tel: (08) 8932 7700 Website: www.ozbrew.com Shop 17 Homemaker Village Millner NT Fax: (08) 8932 7622 Email: [email protected] Before reading these instructions, make sure you have read the information sheet called “Distilling Overview” which is also available in the downloads section of the Oz Brew website. The “Distilling Overview” sheet will provide useful information about the legality of stills and their usages, as well as outlining some good overall basics that will help provide essential information about this process. There are many types of stills, boilers, condensers & carbon filters available on the market. Different products and brands, may use different instructions. These instructions cover one of the most practical methods of distilling using a 25L boiler, a (super) reflux condenser and a Still Spirits Z- carbon filter, which we believe is one of the most effective value for money packages available. Read through all of your instructions including the Helpful Hints in each section before proceeding through the steps in each process. This will ensure you are aware of any tips that may make the process 25 Litre Boiling Pot Z Carbon Filter Super Reflux Condenser easier for you. (3 in 1 Fermenter) Typically produces a high yield of approximately 4-5L @ 80% - 90% V. Stage 1 - Sterilisation 1. Everything used in the fermentation process must be cleaned and sterilised before and after use. This includes the 3 in 1 fermenter, airlock and stirrer. 2. For optimum cleaning, we recommend Combo Cleaner Washing / Steriliser. Wash through all of your equipment with this solution and rinse thoroughly with cold tap water. -

Fractional Distillation for the Laboratory and Industry
ll distillations attempt to separate a lower boiling material (A) from a higher boiling material (B). Packed columns are “packed” A with a material that creates a large surface area within the distillation column. Vapors rise through the column, are condensed by the condenser and fall back down the column. This material is called the condensate. The descending condensate “wets” the packing. Ascending vapors rise through the wet packing and are forced into intimate contact with the condensate. This intimate contact causes the vapor to become enriched in the lower boiling material. This process is often called rectification. A A A AA AA A AA A A AA A A A AA A AA A B/R Instrument Corporation B/R Instrument was founded in 1966 by Roger Roark, Sr. and B A A A A A A B B Harry Brown, Jr. Originally a manufacturer of general glass- A A A AA A A B A A AA AA ware, the company began to specialize in the manufacture B B A A A A A AA AA A A A of spinning band distillation systems in 1968 under a DuPont B A B B B B patent. Since then the company has grown steadily in the A A A A distillation and laboratory equipment markets and now A B A B A B A A A AA distributes products to customers throughout the world. A B A A AA A A A B B A AA A Located in Easton, Maryland on the beautiful Eastern B A A A A A A A B A AA B A Shore of the Chesapeake Bay, the B/R facility encompasses A AA © 2004, B/R Instrument Corporation B A B AA A 14,700 square feet (1,370 square meters) and houses manu- B B A B B A B B A A B A A B A A B A A facturing, sales, engineering, service and administrative A A A A A B B A divisions. -

Building a Still at Low Cost Riku
Building a still at low cost Riku Building a still at low cost Practical guide for building gadgets + few new ideas. Version 0.7-02.02.2005 By Riku – [email protected] v0.7-02.02.2005 web download 1 Building a still at low cost Riku Introduction....................................................................................................................................3 A still..............................................................................................................................................4 How to make a cheap boiler............................................................................................................5 Pot still heads/condensers................................................................................................................9 Water-cooled:..............................................................................................................................9 Air-cooled:................................................................................................................................12 Reflux stills...................................................................................................................................14 Columns....................................................................................................................................14 Size of a column – size does matter...........................................................................................14 Packing.....................................................................................................................................15 -

Reflux Condenser Buyer's Guide
Premium Laboratory Equipment Reflux Condenser Buyer’s Guide Understanding the ① Why refluxing? Air cooled: Reflux condensers with air co- ④ Water consumption and co- oling are available in different versions. In oling costs. Refluxing is one of the most common wor- low-priced systems, heat exchange with air cooled reflux system king methods for organic synthesis. Many the ambient air takes place only on the Extrapolated, an average of 150 liters of chemical reactions only take place under outer surfaces of the glass column. Thus, water flow through a water cooled reflux heat or are accelerated by an increased re- the column must be accordingly high to condenser per hour (Fig. 2 Diagram Water action temperature. Reflux condensers are be able to dissipate heat to the environ- consumption in comparison). Fortunately, used to prevent the solvent of a reaction ment. The much more elaborately desig- many laboratories have installed recircula- Increased requirements on safety and sustainability in the mixture from evaporating even when it is ned reflux condenser system Findenser™ ting chillers or central water recirculating chemical laboratory have been the trigger for the heated for extended periods of time. is equipped with a finned aluminum jacket, systems to reduce the enormous water consumption during refluxing. However, development of new reflux condensers without water which multiplies the heat exchanger surfa- ② Air cooled or water cooled. ce in a compact way. even cooling the coolant has a negative cooling. With the Findenser™, Heidolph offers an What is the difference? impact on the energy balance. efficient, air cooled reflux system, too. The refluxing principle is as simple as it is What are the advantages and disadvantages of reflux ingenious: The boiling vapor from the so- ③ Safety in the laboratory has lution in the flask rises vertically upwards condensers? What are the general aspects to consider many facets. -

Chemical Laboratory Techniques - Gelosa D
FUNDAMENTALS OF CHEMISTRY – Vol. I - Chemical Laboratory Techniques - Gelosa D. and Sliepcevich A. CHEMICAL LABORATORY TECHNIQUES Gelosa D. and Sliepcevich A. Politecnico di Milano, Italy Keywords: batch reactor, boiling point, boiling stones, bubble-plate columns, continuous reactor, cooling bath, crystallization, drying, electric hot plate, electric mantle, equipment, extraction, filtration, fractional distillation, glass joint, glassware, heating bath, laboratory, nonpolar solvents, packed columns, polar solvents, purification, separation, simple distillation, steam distillation, stirring, technique, tubular reactor, vacuum distillation, vacuum pump. Contents 1. Common Laboratory Apparatus 1.1. Glassware 1.2. Heating of the Reaction Mixture 1.2.1. Burners 1.2.2. Heating Baths 1.2.3. Electric Hot Plates and Electric Heating Mantles 1.3. Cooling of the Reaction Mixtures 1.4. Stirring 2. The Reaction 2.1. Batch Reactors 2.2. Continuous Stirred-Tank Reactors 2.3. Tubular Reactors 2.3.1. Pulse Reactors 3. Isolation and Purification Techniques 3.1. Filtration 3.2. Extraction 3.2.1. Liquid–Liquid Extraction 3.2.2. Liquid–Solid Extraction 3.3. Crystallization 3.4. Distillation 3.4.1. TheoreticalUNESCO Aspects – EOLSS 3.4.2. Simple Distillation 3.4.3. Fractional Distillation 3.4.4. Distillation under Reduced Pressure 3.4.5. Steam DistillationSAMPLE CHAPTERS Glossary Bibliography Biographical Sketches Summary Basic laboratory techniques in chemistry have an essential place in the training of a chemist. They provide a good background for experimental skills and for subsequent ©Encyclopedia of Life Support Systems (EOLSS) FUNDAMENTALS OF CHEMISTRY – Vol. I - Chemical Laboratory Techniques - Gelosa D. and Sliepcevich A. independent research. In this chapter, first of all, we shall consider some of the standard pieces of glassware and equipment that you will use in the laboratory. -

Working Principle of Soxhlet Apparatus Pdf
Working principle of soxhlet apparatus pdf Continue The Soxhlet Extractor laboratory is part of a laboratory device invented in 1879 by Franz von Soxhlet. It was originally designed to extract lipids from solid material. Typically, soxhlet extraction is used when the desired compound has a limited solvent solubility, and impurities are insoluble in that solvent. This allows for uncontrolled and restless operation while effectively processing a small amount of solvent to dissolve more material. The description of the Soxhlet extractor has three main sections: the percolator (boiler and reflux), which circulates solvent, thimble (usually from thick filter paper), which retains solid to extract, and a siphon mechanism that periodically empties the thimble. Build the source material containing the compound that will be extracted placed inside the thimble. The thimble is loaded into the main camera of the Soxhlet extractor. The solvent extract to be used is placed in the distillation flask. The flask is placed on the heating element. The Soxhlet extractor is placed on the flask. The reflux capacitor is placed on the extractor. Operation Solvent is heated to reflux. The vapor solvent moves up the distillation arm, and floods into the chamber housing the thimble solid. The capacitor ensures that any vapor solvent is cooled and drips back down into the chamber housing solid material. The camera containing solid material is slowly filled with a warm solvent. Part of the desired compound dissolves into heat solvents. When the Soxhlet camera is almost full, the camera is emptied with a siphon. The solvent returns to the distillation flask. The thimble ensures that the rapid movement of the solvent is not transported by any solid material into a stationary pot.