Download The
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Transport of Dangerous Goods
ST/SG/AC.10/1/Rev.16 (Vol.I) Recommendations on the TRANSPORT OF DANGEROUS GOODS Model Regulations Volume I Sixteenth revised edition UNITED NATIONS New York and Geneva, 2009 NOTE The designations employed and the presentation of the material in this publication do not imply the expression of any opinion whatsoever on the part of the Secretariat of the United Nations concerning the legal status of any country, territory, city or area, or of its authorities, or concerning the delimitation of its frontiers or boundaries. ST/SG/AC.10/1/Rev.16 (Vol.I) Copyright © United Nations, 2009 All rights reserved. No part of this publication may, for sales purposes, be reproduced, stored in a retrieval system or transmitted in any form or by any means, electronic, electrostatic, magnetic tape, mechanical, photocopying or otherwise, without prior permission in writing from the United Nations. UNITED NATIONS Sales No. E.09.VIII.2 ISBN 978-92-1-139136-7 (complete set of two volumes) ISSN 1014-5753 Volumes I and II not to be sold separately FOREWORD The Recommendations on the Transport of Dangerous Goods are addressed to governments and to the international organizations concerned with safety in the transport of dangerous goods. The first version, prepared by the United Nations Economic and Social Council's Committee of Experts on the Transport of Dangerous Goods, was published in 1956 (ST/ECA/43-E/CN.2/170). In response to developments in technology and the changing needs of users, they have been regularly amended and updated at succeeding sessions of the Committee of Experts pursuant to Resolution 645 G (XXIII) of 26 April 1957 of the Economic and Social Council and subsequent resolutions. -

The Oxidizing Behavior of Some Platinum Metal Fluorides
Lawrence Berkeley National Laboratory Lawrence Berkeley National Laboratory Title THE OXIDIZING BEHAVIOR OF SOME PLATINUM METAL FLUORIDES Permalink https://escholarship.org/uc/item/46b5r4v5 Author Graham, Lionell Publication Date 1978-10-01 eScholarship.org Powered by the California Digital Library University of California LBL-8088.c,y '': ·,~, . ' '~~.. .· J!' ... THE OXIDIZING BEHAV!OROF SOME PLATINUM METAL FLUORIDES . Li onell Graham. · (Ph~ D. Thesis ) IIi !l1/ "~ · r-,> ·1cr7o October 1978 ~t .. ~, ~L l ... ;,() · .. t .. J r.~~ r: !\ ~;~, ·l .• ~~:J"~~ t~J ;,~.)(·_ .. {·~ tJ 1v~ ;;:: r~- J··r·r,;~ Ei:~-:·~,:~~ <'11 ().i\{ Prep~red for the .. U. S. Department of Energy. under Contract W~7405~ENG-48 TWO-WEEK LOAN COPY This is a Library Circulating Copy which may be borrowed for two weeks. .. ' ·t•·! •. ·. For a personal retention copy, call Tech. Info. Diu is ion, Ext. 6782 .. LEGAL NOTICE -___,..-----~__, This report was prepared as an account of work sponsored by the United States Government Neither the United States nor the Depart ment of Energy, nor any of their employees, nor any of their con tractors, subcontractors, or their employees, makes any warraf')ty, express or implied,orassumes any legal liabii bility for the accuracy, completeness or usefulness of nformation, appa- ratus, product. ·· · · disclosed; or rep ·. ts that its use would. not infringe p. · · owned rights ... -i- THE OXIDIZING BEHAVIOR OF SOME PLATINUM METAL FLUORIDES Contents Abstract . v I. General Introduction 1 II. General Apparatus and Handling Techniques 2 A. Apparatus . 2 1. General 2 2. The Cryogenic Technology, Inc., Model 21 Cryo-Cooler .. 3 3. Reactor for Gas-Gas Reaction Products 5 B. -

WO 2016/074683 Al 19 May 2016 (19.05.2016) W P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2016/074683 Al 19 May 2016 (19.05.2016) W P O P C T (51) International Patent Classification: (81) Designated States (unless otherwise indicated, for every C12N 15/10 (2006.01) kind of national protection available): AE, AG, AL, AM, AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, (21) International Application Number: BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, PCT/DK20 15/050343 DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, (22) International Filing Date: HN, HR, HU, ID, IL, IN, IR, IS, JP, KE, KG, KN, KP, KR, 11 November 2015 ( 11. 1 1.2015) KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, OM, (25) Filing Language: English PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, SC, (26) Publication Language: English SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (30) Priority Data: PA 2014 00655 11 November 2014 ( 11. 1 1.2014) DK (84) Designated States (unless otherwise indicated, for every 62/077,933 11 November 2014 ( 11. 11.2014) US kind of regional protection available): ARIPO (BW, GH, 62/202,3 18 7 August 2015 (07.08.2015) US GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, ST, SZ, TZ, UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, RU, (71) Applicant: LUNDORF PEDERSEN MATERIALS APS TJ, TM), European (AL, AT, BE, BG, CH, CY, CZ, DE, [DK/DK]; Nordvej 16 B, Himmelev, DK-4000 Roskilde DK, EE, ES, FI, FR, GB, GR, HR, HU, IE, IS, IT, LT, LU, (DK). -

Proceedings of the Nuclear Energy Agency International Workshop on Chemical Hazards in Fuel Cycle Facilities Nuclear Processing
Nuclear Safety NEA/ CSNI/R(2019)9/ADD1 May 2019 www.oecd-nea.org Proceedings of the Nuclear Energy Agency International Workshop on Chemical Hazards in Fuel Cycle Facilities Nuclear Processing Appendix C NEA/CSNI/R(2019)9/ADD1 The non-radiological risks involving dangerous chemicals in nuclear fuel cycle facilities - French framework regulation - Youcef HEMIMOU, Brice DELIME, Raffaello AMOROSI, Josquin VERNON French Nuclear Safety Authority 15, rue Louis Lejeune CS70013 – 92541 Montrouge Cedex [email protected] Accidents involving hazardous chemicals pose a significant threat to the population and the environment. Among nuclear facilities, this threat applies in particular to the fuel cycle facilities. As a consequence, the activities related to hazardous chemicals are covered by a legal framework that, depending on the nature of the activity and the associated risks, aims to guarantee that, they will not be likely to be detrimental to safety, public health and environment [1]. The aim of this article is to present a summary of the main regulations involving dangerous chemicals in nuclear fuel cycle facilities that have to be taken into account in the safety demonstration. The latter must prove that the risks of an accident - radiological or not - and the scale of its consequences, given the current state of knowledge, practices and the vulnerability of the installation environment, are as low as possible under acceptable economic conditions. Key words : nuclear fuel cycle facilities, safety demonstration, principle of defence in depth, deterministic approach, probabilistic approach, dangerous chemicals, non-radiological risks, Seveso Directive, domino effects. 1. THE DANGEROUS SUBSTANCES IN NUCLEAR FUEL CYCLE FACILITIES 1.1. -

List of Lists
United States Office of Solid Waste EPA 550-B-10-001 Environmental Protection and Emergency Response May 2010 Agency www.epa.gov/emergencies LIST OF LISTS Consolidated List of Chemicals Subject to the Emergency Planning and Community Right- To-Know Act (EPCRA), Comprehensive Environmental Response, Compensation and Liability Act (CERCLA) and Section 112(r) of the Clean Air Act • EPCRA Section 302 Extremely Hazardous Substances • CERCLA Hazardous Substances • EPCRA Section 313 Toxic Chemicals • CAA 112(r) Regulated Chemicals For Accidental Release Prevention Office of Emergency Management This page intentionally left blank. TABLE OF CONTENTS Page Introduction................................................................................................................................................ i List of Lists – Conslidated List of Chemicals (by CAS #) Subject to the Emergency Planning and Community Right-to-Know Act (EPCRA), Comprehensive Environmental Response, Compensation and Liability Act (CERCLA) and Section 112(r) of the Clean Air Act ................................................. 1 Appendix A: Alphabetical Listing of Consolidated List ..................................................................... A-1 Appendix B: Radionuclides Listed Under CERCLA .......................................................................... B-1 Appendix C: RCRA Waste Streams and Unlisted Hazardous Wastes................................................ C-1 This page intentionally left blank. LIST OF LISTS Consolidated List of Chemicals -

Chemical Names and CAS Numbers Final
Chemical Abstract Chemical Formula Chemical Name Service (CAS) Number C3H8O 1‐propanol C4H7BrO2 2‐bromobutyric acid 80‐58‐0 GeH3COOH 2‐germaacetic acid C4H10 2‐methylpropane 75‐28‐5 C3H8O 2‐propanol 67‐63‐0 C6H10O3 4‐acetylbutyric acid 448671 C4H7BrO2 4‐bromobutyric acid 2623‐87‐2 CH3CHO acetaldehyde CH3CONH2 acetamide C8H9NO2 acetaminophen 103‐90‐2 − C2H3O2 acetate ion − CH3COO acetate ion C2H4O2 acetic acid 64‐19‐7 CH3COOH acetic acid (CH3)2CO acetone CH3COCl acetyl chloride C2H2 acetylene 74‐86‐2 HCCH acetylene C9H8O4 acetylsalicylic acid 50‐78‐2 H2C(CH)CN acrylonitrile C3H7NO2 Ala C3H7NO2 alanine 56‐41‐7 NaAlSi3O3 albite AlSb aluminium antimonide 25152‐52‐7 AlAs aluminium arsenide 22831‐42‐1 AlBO2 aluminium borate 61279‐70‐7 AlBO aluminium boron oxide 12041‐48‐4 AlBr3 aluminium bromide 7727‐15‐3 AlBr3•6H2O aluminium bromide hexahydrate 2149397 AlCl4Cs aluminium caesium tetrachloride 17992‐03‐9 AlCl3 aluminium chloride (anhydrous) 7446‐70‐0 AlCl3•6H2O aluminium chloride hexahydrate 7784‐13‐6 AlClO aluminium chloride oxide 13596‐11‐7 AlB2 aluminium diboride 12041‐50‐8 AlF2 aluminium difluoride 13569‐23‐8 AlF2O aluminium difluoride oxide 38344‐66‐0 AlB12 aluminium dodecaboride 12041‐54‐2 Al2F6 aluminium fluoride 17949‐86‐9 AlF3 aluminium fluoride 7784‐18‐1 Al(CHO2)3 aluminium formate 7360‐53‐4 1 of 75 Chemical Abstract Chemical Formula Chemical Name Service (CAS) Number Al(OH)3 aluminium hydroxide 21645‐51‐2 Al2I6 aluminium iodide 18898‐35‐6 AlI3 aluminium iodide 7784‐23‐8 AlBr aluminium monobromide 22359‐97‐3 AlCl aluminium monochloride -

Qt5km53474.Pdf
Lawrence Berkeley National Laboratory Recent Work Title REDOX REACTIONS IN THE XeF2/PLATINUM FLUORIDE and XeF2/PALLADIUM FLUORIDE SYSTEMS and THE CONVERSION OF XeF2 T0 XeF4 and Xe Permalink https://escholarship.org/uc/item/5km53474 Author Bartlett, Neil Publication Date 1975-10-01 eScholarship.org Powered by the California Digital Library University of California Submitted to Journal of Fluorine LBL-4195 Chemistry Preprint c.\ REDOX REACTIONS IN THE XeF 2/PLA TINUM FLUORIDE AND XeFz/ PALLADIUM FLUORIDE SYSTEMS AND THE CONVERSION OF XeF 2 TO XeF 4 AND Xe Neil Bartlett, Boris Zemva and Lionell Graham October 1975 ) Prepared for the U. S. Energy Research and Development Administration under Contract W -7405-ENG-48 · For Reference Not to be taken from this room t"' tJ:j t"' I I"\ ,j:>. ....... -..D -- (J'1 r- r r·· r r. n DISCLAIMER This document was prepared as an account of work sponsored by the United States Government. While this document is believed to contain correct information, neither the United States Government nor any agency thereof, nor the Regents of the University of California, nor any of their employees, makes any warranty, express or implied, or assumes any legal responsibility for the accuracy, completeness, or usefulness of any information, apparatus, product, or process disclosed, or represents that its use would not infringe privately owned rights. Reference herein to any specific commercial product, process, or service by its trade name, trademark, manufacturer, or otherwise, does not necessarily constitute or imply its endorsement, recommendation, or favoring by the United States Government or any agency thereof, or the Regents of the University of California. -

Reducing Surface Recombination Velocity in Thin Perovskite Films
Thermochemistry of Ruthenium in Molten Fluoride Salts Metal or Melt? by Laurens G. Braskamp in partial fulfillment of the requirements for the degree of Bachelor of Science in Molecular Science and Technology at the Delft University of Technology to be defended publicly on Monday, August 20, 2018 at 14:30 Student numbers: 4411064 (TU Delft) 1517988 (Universiteit Leiden) Thesis Committee: dr. Elisa Capelli (supervisor) TU Delft dr. Anna L. Smith prof. dr. Rudy J.M. Konings Abstract The Molten Salt Reactor (MSR) is a member of safer and more sustainable fourth generation of nuclear reactors, the development of the might be crucial to continually secure supply of electrical energy from the near future onwards. Because the fluorides of ruthenium (44Ru) are a poorly investigated group of fission products that might be born during MSR operation, some computational research was done on their thermochemistry. Because little experimental data on these species has been obtained, the gas-phase molecular geometries and formation enthalpies of all known Ru-F binary compounds, RuF, RuF2, RuF3, RuF4, RuF5, RuF6, (RuF5)2 and (RuF5)3 are calculated by Density Functional Theory. The DFT/B3LYP method was employed with quasi-relativistic ECP28MWB pseudopotentials and basis set on ruthenium and cc-pVTZ basis set on fluorine. Standard molar entropies and heat capacities were then also calculated by statistical mechanics computations. From the obtained thermochemical data, part of which had to be optimized, and additional data on the solid and liquid phases, the Ru-F subsystem was assessed using the CALPHAD methodology. Based on the developed model, a phase diagram, Ellingham diagram and gas-phase equilibrium diagram were calculated. -

· for Reference
Submitted to Journal of Fluorine LBL-4195 Chemistry Preprint c.\ REDOX REACTIONS IN THE XeF 2/PLA TINUM FLUORIDE AND XeFz/ PALLADIUM FLUORIDE SYSTEMS AND THE CONVERSION OF XeF 2 TO XeF 4 AND Xe Neil Bartlett, Boris Zemva and Lionell Graham October 1975 ) Prepared for the U. S. Energy Research and Development Administration under Contract W -7405-ENG-48 · For Reference Not to be taken from this room t"' tJ:j t"' I I"\ ,j:>. ....... -..D -- (J'1 r- r r·· r r. n DISCLAIMER This document was prepared as an account of work sponsored by the United States Government. While this document is believed to contain correct information, neither the United States Government nor any agency thereof, nor the Regents of the University of California, nor any of their employees, makes any warranty, express or implied, or assumes any legal responsibility for the accuracy, completeness, or usefulness of any information, apparatus, product, or process disclosed, or represents that its use would not infringe privately owned rights. Reference herein to any specific commercial product, process, or service by its trade name, trademark, manufacturer, or otherwise, does not necessarily constitute or imply its endorsement, recommendation, or favoring by the United States Government or any agency thereof, or the Regents of the University of California. The views and opinions of authors expressed herein do not necessarily state or reflect those of the United States Government or any agency thereof or the Regents of the University of California. -1- REDOX REACTIONS IN THE XeFz/PLATINUM FLUORIDE AND XeFz/PALLADIID-1 FLUORIDE SYSTEMS AND THE CONVERSION OF XeFz TO XeF4 AND Xe \J NEIL BARTLETT, BORIS ZEMVA AND LIONELL GRAHAJ1 Department of Chemistry, University of California, and Inorganic Haterials Research Division, Lawrence Berkeley Laboratory, Berkeley, California 94720 (U.S.A.) (To Professor George H. -
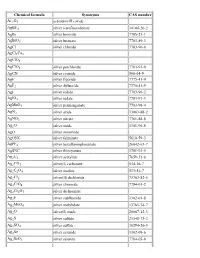
List of Chemical Formulas
Chemical formula Synonyms CAS number Ac2O3 actinium(III) oxide AgBF4 silver tetrafluoroborate 14104-20-2 AgBr silver bromide 7785-23-1 AgBrO3 silver bromate 7783-89-3 AgCl silver chloride 7783-90-6 AgCl3Cu2 AgClO3 AgClO4 silver perchlorate 7783-93-9 AgCN silver cyanide 506-64-9 AgF silver fluoride 7775-41-9 AgF2 silver difluoride 7775-41-9 AgI silver iodide 7783-96-2 AgIO3 silver iodate 7783-97-3 AgMnO4 silver permanganate 7783-98-4 AgN3 silver azide 13863-88-2 AgNO3 silver nitrate 7761-88-8 Ag2O silver oxide 1301-96-8 AgO silver monoxide AgONC silver fulminate 5610-59-3 AgPF6 silver hexafluorophosphate 26042-63-7 AgSNC silver thiocyanate 1701-93-5 Ag2C2 silver acetylide 7659-31-6 Ag2CO3 silver(I) carbonate 534-16-7 Ag2C2O4 silver oxalate 533-51-7 Ag2Cl2 silver(II) dichloride 75763-82-5 Ag2CrO4 silver chromate 7784-01-2 Ag2Cr2O7 silver dichromate Ag2F silver subfluoride 1302-01-8 Ag2MoO4 silver molybdate 13765-74-7 Ag2O silver(I) oxide 20667-12-3 Ag2S silver sulfide 21548-73-2 Ag2SO4 silver sulfate 10294-26-5 Ag2Se silver selenide 1302-09-6 Ag2SeO3 silver selenite 7784-05-6 Ag2SeO4 silver selenate 7784-07-8 Ag2Te silver(I) telluride 12002-99-2 Ag3Br2 silver dibromide 11078-32-3 Ag3Br3 silver tribromide 11078-33-4 Ag3Cl3 silver(III) trichloride 12444-96-1 Ag3I3 silver(III) triiodide 37375-12-5 Ag3PO4 silver phosphate 7784-09-0 AlBO aluminium boron oxide 12041-48-4 AlBO2 aluminium borate 61279-70-7 AlBr aluminium monobromide 22359-97-3 AlBr3 aluminium tribromide 7727-15-3 AlCl aluminium monochloride 13595-81-8 AlClF aluminium chloride fluoride -

THE FLUORIDES of PLATINUM and R E L a T E D Compounds by DEREK HARRY LOHMANN B.Sc, University of London, 1953 M.Sc, Queen's Univ
THE FLUORIDES OF PLATINUM and related compounds by DEREK HARRY LOHMANN B.Sc, University of London, 1953 M.Sc, Queen's University, Ontario, 1959 A THESIS SUBMITTED IN PARTIAL FULFILMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY in the Department of CHEMISTRY We accept this thesis as conforming to the required standard. THE UNIVERSITY OF BRITISH COLUMBIA October 1961, In presenting this thesis in partial fulfilment of the requirements for an advanced degree at the University of British Columbia, I agree that the Library shall make it freely available for reference and study. I further agree that- permission for extensive copying of this thesis for scholarly purposes may be granted by the Head of my Department or by his representatives. It is understood that copying or publication of this thesis for financial gain shall not be allowed without my written permission. Department of CHEMISTRY The University of British Columbia, Vancouver 8, Canada. Date 31st October 1961. %\\t Pntesttg of ^irtttslj Columbia FACULTY OF GRADUATE STUDIES PROGRAMME OF THE FINAL ORAL EXAMINATION FOR THE DEGREE OF DOCTOR OF PHILOSOPHY PUBLICATIONS oi . I. "Polar effects in Hydrogen abstraction reactions," M. P. Godsay, DEREK HARRY LOHMANN D. H. Lohmann and K. E. Russell, Chem. and Ind., 1959, 1603. B.Sc, University of London, 1953 M.Sc, Queen's University, Ontario, 1959 2. "Two new fluorides of Platinum," N. Bartlett and D. H. Lohmann, Proc. Chem. Soc, 1960, 14. TUESDAY, NOVEMBER 28th, 1961, AT 1:00 P.M. 3. "The reaction of 2,2-Diphenyl-l-Picrylhydrazyl with 9,10-Dihy- droanthracene and 1,4-Dihydronaphthalene," I. -

Nomenclature of Inorganic Chemistry (IUPAC Recommendations 2005)
NOMENCLATURE OF INORGANIC CHEMISTRY IUPAC Recommendations 2005 IUPAC Periodic Table of the Elements 118 1 2 21314151617 H He 3 4 5 6 7 8 9 10 Li Be B C N O F Ne 11 12 13 14 15 16 17 18 3456 78910 11 12 Na Mg Al Si P S Cl Ar 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 K Ca Sc Ti V Cr Mn Fe Co Ni Cu Zn Ga Ge As Se Br Kr 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 Rb Sr Y Zr Nb Mo Tc Ru Rh Pd Ag Cd In Sn Sb Te I Xe 55 56 * 57− 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 Cs Ba lanthanoids Hf Ta W Re Os Ir Pt Au Hg Tl Pb Bi Po At Rn 87 88 ‡ 89− 103 104 105 106 107 108 109 110 111 112 113 114 115 116 117 118 Fr Ra actinoids Rf Db Sg Bh Hs Mt Ds Rg Uub Uut Uuq Uup Uuh Uus Uuo * 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 La Ce Pr Nd Pm Sm Eu Gd Tb Dy Ho Er Tm Yb Lu ‡ 89 90 91 92 93 94 95 96 97 98 99 100 101 102 103 Ac Th Pa U Np Pu Am Cm Bk Cf Es Fm Md No Lr International Union of Pure and Applied Chemistry Nomenclature of Inorganic Chemistry IUPAC RECOMMENDATIONS 2005 Issued by the Division of Chemical Nomenclature and Structure Representation in collaboration with the Division of Inorganic Chemistry Prepared for publication by Neil G.