Does the Pharmacology of Oxycodone Justify Its Increasing Use As an Analgesic?
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Metamorphosis of Hydromorphone Gary M
Pharmacy PersPective The metamorphosis of hydromorphone Gary M. Reisfield, MD George R. Wilson, MD introduction “That’s it. The Mayo Clinic at Rochester devel - oped it, the word and the drug, for it means a Hydromorphone hydrochloride, one of the oldest of drug, a pain relieving drug, five times as potent the extant opioid analgesics, has been in clinical use for as morphine, as harmless as water and with no more than 70 years. Its use by the oral route in chronic habit forming qualities. pain and hospice/palliative medicine settings has been limited, however, largely owing to its relatively short “The medical journals say it is particularly useful duration of action. With the recent US Food and Drug in the operation of cases where other drugs Administration (FDA) approval of a once-daily extended- seem to offer no relief from pain. Unlike mor - release formulation of the drug (Palladone, Purdue phine, there are no pleasurable sensations to its Pharma LP, Stamford, CT), hydromorphone joins mor - use, however, and if the doctors reckon correctly phine, oxycodone, and fentanyl as the only extended- its use may go far toward curing addicts of the release opioids available on the United States market. morphine habit.” Here, we review the history, pharmacokinetics, and other relevant issues concerning this invaluable opioid, and Montgomery (AL) Advertiser , Dec. 18, 1932 also discuss the role of the new formulation in the man - agement of chronic pain. From 1929 to 1939, the National Research Council’s Committee on Drug Addiction conducted exhaustive history research on the morphine molecule and its analogs, pro - ducing more than 150 semisynthetic and more than 300 Hydromorphone [also Dilaudid (Knoll Laboratories, synthetic compounds, of which more than 30 were tested Mount Olive, NJ), dihydromorphinone, dihydromorphe - clinically. -

Morphinone Reductase for the Preparation Of
Europäisches Patentamt *EP000649465B1* (19) European Patent Office Office européen des brevets (11) EP 0 649 465 B1 (12) EUROPEAN PATENT SPECIFICATION (45) Date of publication and mention (51) Int Cl.7: C12N 9/02, C12P 17/18, of the grant of the patent: C12Q 1/32 24.07.2002 Bulletin 2002/30 (86) International application number: (21) Application number: 93913236.1 PCT/GB93/01129 (22) Date of filing: 28.05.1993 (87) International publication number: WO 94/00565 (06.01.1994 Gazette 1994/02) (54) MORPHINONE REDUCTASE FOR THE PREPARATION OF HYDROMORPHONE AND HYDROCODONE MORPHINONE REDUKTASE ZUR HERSTELLUNG VON HYDROMORPHONE UND HYDROCODONE MORPHINONE REDUCTASE DESTINEE A LA PREPARATION D’HYDROMORPHONE ET D’HYDROCODONE (84) Designated Contracting States: (74) Representative: Perry, Robert Edward AT BE CH DE DK ES FR GB IE IT LI NL PT SE GILL JENNINGS & EVERY Broadgate House (30) Priority: 25.06.1992 GB 9213524 7 Eldon Street London EC2M 7LH (GB) (43) Date of publication of application: 26.04.1995 Bulletin 1995/17 (56) References cited: WO-A-90/13634 (73) Proprietor: MACFARLAN SMITH LIMITED Edinburgh EH11 2QA (GB) • THE BIOCHEMICAL JOURNAL vol. 274, no. 3, 15 March 1991, pages 875 - 880 NEIL C. BRUCE ET (72) Inventors: AL. ’Microbial degradation of the morphine • HAILES, Anne, Maria 59 Lower Road alkaloids. Purification and characterization of Kent DA8 1AY (GB) morphine dehydrogenase from Pseudomonas • FRENCH, Christopher, Edward putida M10’ Palmerston North (NZ) • BRUCE, Neil, Charles Cambridge CB2 1ND (GB) Note: Within nine months from the publication of the mention of the grant of the European patent, any person may give notice to the European Patent Office of opposition to the European patent granted. -

Microbial Degradation of the Morphine Alkaloids Purification and Characterization of Morphine Dehydrogenase from Pseudomonas Putida M10
Biochem. J. (1991) 274, 875-880 (Printed in Great Britain) 875 Microbial degradation of the morphine alkaloids Purification and characterization of morphine dehydrogenase from Pseudomonas putida M10 Neil C. BRUCE,* Clare J. WILMOT, Keith N. JORDAN, Lauren D. Gray STEPHENS and Christopher R. LOWE Institute of Biotechnology, University of Cambridge, Tennis Court Road, Cambridge CB2 1QT, U.K. The NADP+-dependent morphine dehydrogenase that catalyses the oxidation of morphine to morphinone was detected in glucose-grown cells of Pseudomonas putida M 10. A rapid and reliable purification procedure involving two consecutive affinity chromatography steps on immobilized dyes was developed for purifying the enzyme 1216-fold to electrophoretic homogeneity from P. putida M 10. Morphine dehydrogenase was found to be a monomer of Mr 32000 and highly specific with regard to substrates, oxidizing only the C-6 hydroxy group of morphine and codeine. The pH optimum of morphine dehydrogenase was 9.5, and at pH 6.5 in the presence of NADPH the enzyme catalyses the reduction of codeinone to codeine. The Km values for morphine and codeine were 0.46 mm and 0.044 mm respectively. The enzyme was inhibited by thiol-blocking reagents and the metal-complexing reagents 1, 10-phenanthroline and 2,2'-dipyridyl, suggesting that a metal centre may be necessary for activity of the enzyme. INTRODUCTION EXPERIMENTAL The morphine alkaloids have attracted considerable attention Materials owing to their analgaesic properties and, consequently, much Mimetic Orange 3 A6XL and Mimetic Red A6XL were effort in the past has been directed at the production of new obtained from Affinity Chromatography Ltd., Freeport, morphine alkaloids by micro-organisms (lizuka et al., 1960, Ballasalla, Isle of Man, U.K. -
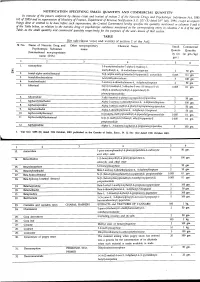
NOTIFICATION SPECIFYING SMALL QUANTITY and COMMERCIAL QUANTITYI Ly of the Pozoersconferred (Xxiiiil
NOTIFICATION SPECIFYING SMALL QUANTITY AND COMMERCIAL QUANTITYI ly of the pozoersconferred (xxiiiil . - lleyise by clansesfuiia) and of section2 of the Nnrcotic Drrtgsnnd pnlchotropic SrtbstancesAct, 19g5 (61 of 1985)and in supersessionof Ministry of Ftnnnce, (D Departmentof'Reuenue Noiification s.o.527 aotra i'6tt,1uli, tssi, exceptas respects things doneor omitted to be done .beforesuch supersession,the Cential Gooernmenihereby spectfiesthe qtnntitrl ,nlnt'ionedin coltnnnsS and 6 of the Tablebelow, in relationto.the narcoticdrug or psychotropicsubstnnce mentioned.ii tlr', ,1urrponding entnl in columns2 to 4 of thesaid Table,as the small quantitrl nnd commercialquantity respectiailyfor the purposesof the said clnuse:sof ttrit seciion. TABLE [Seesub-clause vii(a) and xxiii(a) of section 2 of the Act] Sl No. Name of Narcotic Drug and Other non-proprietary Chemical Name Small Commercial Psychotropic Substance name Quanti- Quantity (Intemational non-proprietary ity (in (in gm./kg.) name (INN) Acetorphine 3-0-acetyltetrahydro-7-alpha-(l-hydroxy-l- methylbutyt)-o, l4-endoetheno-onpavine $ 50 9.. 1\) Acetyl-alpha-methylfen N-[-(alpha-methylphenethyl)-4-piperidy]l acetanilide 0.1 g-. J. Acetyldihydrocodeine 100 4. Acetylmethadol 3-acetoxy-6-dimethylamino-4, 4 lheptane 50 gm. 5. Alfentanil -ethyl-4, N-[1-[2-( 5-dihydro-S-oxo-lH-tetrazol-t-yt) 01 gm. ethyll-4-(methoxymethyl)-4-piperidinyll -N- ylpropanamide Allyprodine 3-allyl Jmethyl-4-phenyl-4 Alpha-3-acetoxy-6-dimethylamino-4,Ld lheptane 100 gm Alpha-3-ethyl-l-methyl-4-phenyl-4-propionox 50 gm. 9.7' AlphamethadolArPnametnaool Alpha-6-dimethylamino-4, 4-diphenyl-3-heptanol 2 S0 gm. 10. Alphu-*"thylf"ntur,yl 11. -

0 Cover Part Two\374
PART TWO DEUXIÈME PARTIE SEGUNDA PARTE ﺍﳉﺰﺀ ﺍﻟﺜﺎﱐ 第二部分 ЧАСТЬ ВТΟΡАЯ - (...), [...] - (-)-3-hidroxi-N- (-)-(2R)-N-méthyl-1- fenacilmorfinán → Levophenacyl-morphan phénylpropan-2-amine → Levometamfetamine (-)-3-hidroxi-N- (-)-(3S,6R)-6- metilmorfinán → Levorphanol (dimethylamino)-4,4- (-)-3-hydroksymorfinan → Norlevorphanol diphenyl-3-heptanol → Betamethadol (-)-3-hydroksy-N- (-)-(3S,6R)-6- fenacylmorfinan → Levophenacyl-morphan (dimethylamino)-4,4- (-)-3-hydroksy-N- diphenyl-3-heptanol metylmorfinan → Levorphanol acetate (ester) → Betacetylmethadol (-)-3-hydroxymorphinan → Norlevorphanol (-)-(5R)-4,5-epoxy-3- (-)-3-hydroxy-N- methoxy-9α-methyl methylmorphinan → Levorphanol → Thebacon morphin-6-en-6-yl acetate (-)-3-hydroxynormorphinan → Norlevorphanol (-)-(5R,6S)-3-benzyloxy-4,5- (-)-3-hydroxy-N- epoxy-9a-methylmorphin- phenacylmorphinan → Levophenacyl-morphan 7-en-6-yl myristate → Myrophine (-)-3-hydroxytropane-2- (-)-(5R,6S)-4,5-epoxy carboxylat → Ecgonine morphin-7-en-3,6-diol → Normorphine (-)-3-idrossi-N- (-)-(5R,6S,14S)-4,5-epoxy- metilmorfinano → Levorphanol 14-hydroxy-3-methoxy- (-)-3-methoxy-N- 9a-methylmorphinan-6- methylmorphinan → Levomethorphan one → Oxycodone (-)-3-metil-2,2-difenil-4- (-)-(R)-4,5-epoxy-3- morfolinbutirilpirrolidina → Levomoramide methoxy-9a-methyl (-)-3-metoksy-N- morphinan-6-one → Hydrocodone metylmorfinan → Levomethorphan (-)-(R)-6-(dimethylamino)- (-)-3-metossi-N-metil- 4,4-diphenyl-3-heptanone → l-methadone morfinano → Levomethorphan (-)-(R)-N,α-dimethyl (-)-3-metoxi-N- phenethylamine → Levometamfetamine -

Ep 2398808 B1
(19) TZZ ¥Z_T (11) EP 2 398 808 B1 (12) EUROPEAN PATENT SPECIFICATION (45) Date of publication and mention (51) Int Cl.: of the grant of the patent: C07D 489/08 (2006.01) 20.11.2013 Bulletin 2013/47 (86) International application number: (21) Application number: 10704301.0 PCT/US2010/024372 (22) Date of filing: 17.02.2010 (87) International publication number: WO 2010/096416 (26.08.2010 Gazette 2010/34) (54) Process for the reductive alkylation of normorphinans VERFAHREN ZUR REDUKTIVEN ALKYLIERUNG VON NORMORPHINANEN Procédé d’alkylation réductrice des normorphinanes (84) Designated Contracting States: • WOODS, Sharon AT BE BG CH CY CZ DE DK EE ES FI FR GB GR Florissant HR HU IE IS IT LI LT LU LV MC MK MT NL NO PL Missouri 63034 (US) PT RO SE SI SK SM TR (74) Representative: Beckmann, Claus (30) Priority: 17.02.2009 US 153021 P Kraus & Weisert Patent- und Rechtsanwälte (43) Date of publication of application: Thomas-Wimmer-Ring 15 28.12.2011 Bulletin 2011/52 80539 München (DE) (73) Proprietor: Mallinckrodt LLC (56) References cited: Hazelwood, MO 63042 (US) WO-A1-2006/035195 WO-A1-2009/012005 (72) Inventors: • HUDSON, Edmund, C. Clayton Missouri 63105 (US) Note: Within nine months of the publication of the mention of the grant of the European patent in the European Patent Bulletin, any person may give notice to the European Patent Office of opposition to that patent, in accordance with the Implementing Regulations. Notice of opposition shall not be deemed to have been filed until the opposition fee has been paid. -

Back Matter (PDF)
The Editor of the Journal of Pharmacology & Experimental Therapeutics wishes to express appreciation to the following colleagues who acted as Guest Editors for Specific Fields in 1987. Martin W. Adler Frank R. Goodman Elaine Sanders-Bush Lewis Aronow Robert Z. Gussin Lewis E. Seiden C. Paul Bianchi Conan Kornetsky Theodore J. Torphy Allan H. Conney Donald J. Jenden John L. Skosey Theodore J. Cicero Robert M. Joy Larry G. Stark Charles R. Craig Louis Lemberger Norman Weiner Gerard L. Gebber Benedict R. Lucchesi Wallace D. Winters James W. Gibb Donald V. Priola Dixon M. Woodbury Dora B. Goldstein Additionally, sincere thanks are due to 724 of our colleagues not on the Editorial Advisory Board who reviewed articles for the Journal in 1987. Their contributions are acknowledged individually in the Annual Report of The Journal to The Board of Publications Trustees of The American Society for Pharmacology and Experimental Therapeutics. INDEX Volume 245, April-June, 1988 A23187, effect ofextracellularcalcium omis- classification, ligand affmity and mo- -induced intoxication, effects of Ro15- sion, hepatocytes (rats), 493 lecular mass (bovine), 1060 4513 (rats), 880 Abhold, R. H. and Harding, J. W.: Metabo- imidazoline analogs, aorta (rats), 793 Mi, S. F., see Schulze, G. E., 178 lism of angiotensins II and III by inotropic positive effect, inositol tris- Alkondon, M., Rao, K. S. and Albuquerque, membrane-bound peptidases from phosphate increase in heart (rats), E. X.: Acetylcholinesterase reactiva- rat brain, 171 327 tom modify the functional properties Abiko, Y., see Hara, Y., 305 prazosin and indoramin, coronary of the nicotinic acetylcholine recep- Acetaminophen blood flow (dogs), 232 tor ion channel, 543 diminuition of hepatotoxicity, ethanol alpha-2 Allbee, W. -

Recent Advances in the Chemistry of Oripavine and Its Derivatives
Advances in Bioscience and Biotechnology, 2014, 5, 704-717 Published Online July 2014 in SciRes. http://www.scirp.org/journal/abb http://dx.doi.org/10.4236/abb.2014.58084 Recent Advances in the Chemistry of Oripavine and Its Derivatives Sandor Hosztafi Institute of Pharmaceutical Chemistry, Semmelweis University, Budapest, Hungary Email: [email protected] Received 2 May 2014; revised 16 June 2014; accepted 12 July 2014 Copyright © 2014 by author and Scientific Research Publishing Inc. This work is licensed under the Creative Commons Attribution International License (CC BY). http://creativecommons.org/licenses/by/4.0/ Abstract Oripavine is the major alkaloid of Papaver orientale. It is an important intermediate in the bio- synthesis of morphine alkaloids. Recently, new Papaver somniferum strains have been developed which accumulate thebaine and oripavine, but not morphine and codeine. Therefore, the chemi- stry of oripavine has been studied intensively to synthesize opioid pharmaceuticals such as oxy- morphone, naloxone and buprenorphine. Keywords Papaver orientale, Papaver somniferum, Oripavine, Thebaine, Top1 Poppy 1. Introduction Thebaine an alkaloid present in 0.2% - 0.8% in opium and a major constituent (90% of total alkaloid content) in Papaver bracteatum (which is morphine free), possesses little utility medically for two reasons: (a) its lack of the depressant and analgesic properties common to other morphine alkaloids and (b) its expression of extreme toxicity and CNS stimulation. Oripavine is also a minor alkaloid of the opium poppy, but it is the main alkaloid of the oriental poppy Papaver orientale, which is a perennial flowering plant. The development of the top1 poppy was an important finding for the opium industry in Australia, because this poppy strain produces thebaine and oripavine, but not morphine or codeine. -

Problems of Drug Dependence 1996: Proceedings of the 58Th Annual Scientific Meeting the College on Problems of Drug Dependence, Inc
National Institute on Drug Abuse RESEARCH MONOGRAPH SERIES Problems of Drug Dependence 1996: Proceedings of the 58th Annual Scientific Meeting The College on Problems of Drug Dependence, Inc. 174 U.S. Department of Health and Human Services • National Institutes of Health Problems of Drug Dependence 1996: Proceedings of the 58th Annual Scientific Meeting The College on Problems of Drug Dependence, Inc. Editor: Louis S. Harris, Ph.D. Virginia Commonwealth University NIDA Research Monograph 174 1997 U.S. DEPARTMENT OF HEALTH AND HUMAN SERVICES National Institutes of Health National Institute on Drug Abuse 5600 Fishers Lane Rockville, MD 20857 BOARD OF DIRECTORS Robert L. Balster, Ph.D., President Thomas R. Kosten, M.D. Steven G. Holtzman, Ph.D., President-Elect Michael J. Kuhar, Ph.D. Edward M. Sellers, M.D., Ph.D., Past-President Scott E. Lukas, Ph.D. George E. Bigelow, Ph.D., Treasurer Billy R. Martin, Ph.D. Edgar H. Brenner, J.D. A. Thomas McLellan, Ph.D. Leonard Cook, Ph.D. Roy W. Pickens, Ph.D. Linda B. Cottler, Ph.D, M.P.H. Beny J. Primm, M.D. Linda A. Dykstra, Ph.D. Peter H. Reuter, Ph.D. Avram Goldstein, M.D. Sidney H. Schnoll, M.D., Ph.D. Charles W. Gorodetzky, M.D., Ph.D. Charles R. Schuster, Ph.D. John R. Hughes, M.D. James E. Smith, Ph.D. M. Ross Johnson, Ph.D. Maxine L. Stitzer, Ph.D. EXECUTIVE OFFICER Martin W. Adler, Ph.D. SCIENTIFIC PROGRAM COMMITTEE Thomas R. Kosten, Chair Martin W. Adler Maureen E. Bronson Kathryn A. Cunningham Ellen B. Geller M. -

Benefits and Risks of Pharmaceutical Opioids
Benefits and risks of pharmaceutical opioids: Essential treatment and diverted medication A global review of availability, extra-medical use, injection and the association with HIV Louisa Degenhardt, Briony Larance, Bradley Mathers, Tasnim Azim, Adeeba Kamarulzaman, Richard Mattick, Samiran Panda, Abdalla Toufik, Mark Tyndall, Lucas Wiessing and Alex Wodak on behalf of the Reference Group to the United Nations on HIV and injecting drug use National Drug and Alcohol Research Centre UNIVERSITY OF NEW SOUTH WALES Sydney, Australia i ISBN: 978-0-7334-2707-7 This work is copyright. You may download, display, print and reproduce this material in unaltered form only (retaining this notice) for your personal, non-commercial use or use within your organisation. All other rights are reserved. Requests and enquiries concerning reproduction and rights should be addressed to the information manager, National Drug and Alcohol Research Centre, University of New South Wales, Sydney, NSW 2052, Australia. ii Acknowledgements This report is a product of the Reference Group to the United Nations on HIV and injecting drug use and was reviewed by the 2007 members of the Reference Group and also the Secretariat of the Reference Group. In 2007 the Reference Group members were Tasnim Azim, Mauro Guarinieri, Matthew Hickman, Adeeba Kamarulzaman, Kasia Malinowska-Sempruch, Fabio Mesquita, Azarakhsh Mokri, Olanrewaju Olusola Onigbogi, Fred Owiti, Samiran Panda, Steffanie A. Strathdee, Fayzal Sulliman, Abdalla Toufik, Jallal Toufiq, Mark Tyndall and Lucas Wiessing. In 2007 the Secretariat consisted of Richard Mattick, Louisa Degenhardt, Bradley Mathers, Benjamin Phillips, Kate Dolan and Alex Wodak. The following individuals assisted with the compilation of the literature: • Laura Kemmis, United Kingdom • Gabrielle Campbell, NDARC, University of NSW • Eva Congreve, NDARC, University of NSW • Benjamin Phillips, NDARC, University of NSW • Jessica Singleton, NDARC. -
Natural Opioid Peptides, Synthetic Analogs, Peptidases: Pharmacological Analysis and Drug Design
SEMMELWEIS UNIVERSITY Doctoral School of Pharmaceutical and Pharmacological Sciences NATURAL OPIOID PEPTIDES, SYNTHETIC ANALOGS, PEPTIDASES: PHARMACOLOGICAL ANALYSIS AND DRUG DESIGN by András Z. Rónai Ph.D. Dissertation Supervisor: Prof. Dr. Kálmán Magyar, D.Sc. Member of Hungarian Academy of Sciences Department of Pharmacology and Pharmacotherapy Semmelweis University, Budapest Budapest, 2004 1 To Sándor Bajusz 2 CONTENTS 1. Introduction 2. List of abbreviations 3. Results and Conclusions 3.1. Chapter 1. Critical overview of the use of isolated organs in opioid research 3.2. Chapter 2. Pharmacological characterisation of enkephalins, b-endorphin and its fragments 3.3. Chapter 3. Development of potent analgesic enkephalin analogs such as (D-Met2,Pro5) -enkephalinamide 3.4. Chapter 4. Characterisation of structural principles for the receptor type-selectivity of enkephalin analogs 3.5. Chapter 5. Detection of a novel dipeptidyl-carboxypeptidase in rodent blood vessel and human adrenomedullary tumors 3.6. Chapter 6. Design and pharmacological characterisation of novel, pure and highly d -opioid receptor type-selective and k-opioid receptor type.selective peptide antagonists: the principle of N-acylation. 3.7. Chapter 7. Present and future: endomorphins, nociceptin. 4. General summary and conclusions 5. Acknowledgement 6. References 7. Patents 8. Appendix. 3 1. Introduction The cornerstone of opioid peptide history was the discovery of enkephalins (Tyr-Gly-Gly- Phe-Met, (Met5) enkephalin and Tyr-Gly-Gly-Phe-Leu, (Leu5)-enkephalin) in porcine brain by Hughes, Kosterlitz and their coworkers in 1975 (53). The history appeared to obey the principle of receptor theory declaring that a receptor should have one or more endogenous ligand(s). -
Electrospray Tandem Mass Spectrometric Investigations of Morphinans
Electrospray Tandem Mass Spectrometric Investigations of Morphinans Klaus Raith and Reinhard Neubert Institute of Pharmaceutics and Biopharmaceutics, Martin Luther University, Halle, Germany Chotima Poeaknapo, Christian Boettcher, and Meinhart H. Zenk Biocenter, Martin Luther University, Halle, Germany Ju¨rgen Schmidt Leibniz Institute of Plant Biochemistry, Halle, Germany ϩ In this study positive ESI tandem mass spectra of the [M ϩ H] ions of morphinan alkaloids obtained using an ion trap MS were compared with those from a triple quadrupole MS. This allows to assess the differences of the tandem-in-time versus the tandem-in-space principle, often hampering the development of ESI MS/MS libraries. Fragmentation pathways and possible fragment ion structures were discussed. In order to obtain elemental composition, accurate mass measurements were performed. According to the MS/MS fragmentation pathway, the investigated compounds can be grouped into 4 subsets: (1) morphine and codeine, (2) morphinone, codeinone, and neopinone, (3) thebaine and oripavine, (4) salutaridine and salutaridinol. Salutaridinol-7-O-acetate shows a different fragmentation behavior because of the favored loss of acetic acid. Although most fragment ions occur in both ion trap and triple quad tandem mass spectra, some are exclusively seen in either type. For triple quad, quadrupole time-of-flight and FT-ICR MS/MS, the base peak of morphine results from an ion at m/z 165 that contains neither nitrogen nor oxygen. This ion is not found in ion trap MS/MS, but in subsequential MS3 and MS4. (J Am Soc Mass Spectrom 2003, 14, 1262–1269) © 2003 American Society for Mass Spectrometry espite the importance of morphine and its to magnetic sector instruments, triple quadrupole, biosynthetic relatives, the literature dealing quadrupole ion trap, and quadrupole time-of-flight Dwith electrospray ionization mass spectro- are all considered to perform low collision energy metry is limited.