Microbial Degradation of the Morphine Alkaloids Purification and Characterization of Morphine Dehydrogenase from Pseudomonas Putida M10
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Metamorphosis of Hydromorphone Gary M
Pharmacy PersPective The metamorphosis of hydromorphone Gary M. Reisfield, MD George R. Wilson, MD introduction “That’s it. The Mayo Clinic at Rochester devel - oped it, the word and the drug, for it means a Hydromorphone hydrochloride, one of the oldest of drug, a pain relieving drug, five times as potent the extant opioid analgesics, has been in clinical use for as morphine, as harmless as water and with no more than 70 years. Its use by the oral route in chronic habit forming qualities. pain and hospice/palliative medicine settings has been limited, however, largely owing to its relatively short “The medical journals say it is particularly useful duration of action. With the recent US Food and Drug in the operation of cases where other drugs Administration (FDA) approval of a once-daily extended- seem to offer no relief from pain. Unlike mor - release formulation of the drug (Palladone, Purdue phine, there are no pleasurable sensations to its Pharma LP, Stamford, CT), hydromorphone joins mor - use, however, and if the doctors reckon correctly phine, oxycodone, and fentanyl as the only extended- its use may go far toward curing addicts of the release opioids available on the United States market. morphine habit.” Here, we review the history, pharmacokinetics, and other relevant issues concerning this invaluable opioid, and Montgomery (AL) Advertiser , Dec. 18, 1932 also discuss the role of the new formulation in the man - agement of chronic pain. From 1929 to 1939, the National Research Council’s Committee on Drug Addiction conducted exhaustive history research on the morphine molecule and its analogs, pro - ducing more than 150 semisynthetic and more than 300 Hydromorphone [also Dilaudid (Knoll Laboratories, synthetic compounds, of which more than 30 were tested Mount Olive, NJ), dihydromorphinone, dihydromorphe - clinically. -

Morphinone Reductase for the Preparation Of
Europäisches Patentamt *EP000649465B1* (19) European Patent Office Office européen des brevets (11) EP 0 649 465 B1 (12) EUROPEAN PATENT SPECIFICATION (45) Date of publication and mention (51) Int Cl.7: C12N 9/02, C12P 17/18, of the grant of the patent: C12Q 1/32 24.07.2002 Bulletin 2002/30 (86) International application number: (21) Application number: 93913236.1 PCT/GB93/01129 (22) Date of filing: 28.05.1993 (87) International publication number: WO 94/00565 (06.01.1994 Gazette 1994/02) (54) MORPHINONE REDUCTASE FOR THE PREPARATION OF HYDROMORPHONE AND HYDROCODONE MORPHINONE REDUKTASE ZUR HERSTELLUNG VON HYDROMORPHONE UND HYDROCODONE MORPHINONE REDUCTASE DESTINEE A LA PREPARATION D’HYDROMORPHONE ET D’HYDROCODONE (84) Designated Contracting States: (74) Representative: Perry, Robert Edward AT BE CH DE DK ES FR GB IE IT LI NL PT SE GILL JENNINGS & EVERY Broadgate House (30) Priority: 25.06.1992 GB 9213524 7 Eldon Street London EC2M 7LH (GB) (43) Date of publication of application: 26.04.1995 Bulletin 1995/17 (56) References cited: WO-A-90/13634 (73) Proprietor: MACFARLAN SMITH LIMITED Edinburgh EH11 2QA (GB) • THE BIOCHEMICAL JOURNAL vol. 274, no. 3, 15 March 1991, pages 875 - 880 NEIL C. BRUCE ET (72) Inventors: AL. ’Microbial degradation of the morphine • HAILES, Anne, Maria 59 Lower Road alkaloids. Purification and characterization of Kent DA8 1AY (GB) morphine dehydrogenase from Pseudomonas • FRENCH, Christopher, Edward putida M10’ Palmerston North (NZ) • BRUCE, Neil, Charles Cambridge CB2 1ND (GB) Note: Within nine months from the publication of the mention of the grant of the European patent, any person may give notice to the European Patent Office of opposition to the European patent granted. -

Quinic Acid-Mediated Induction of Hypovirulence and a Hypovirulence-Associated Double-Stranded RNA in Rhizoctonia Solani Chunyu Liu
The University of Maine DigitalCommons@UMaine Electronic Theses and Dissertations Fogler Library 8-2001 Quinic Acid-Mediated Induction of Hypovirulence and a Hypovirulence-Associated Double-Stranded RNA in Rhizoctonia Solani Chunyu Liu Follow this and additional works at: http://digitalcommons.library.umaine.edu/etd Part of the Biochemistry Commons, and the Molecular Biology Commons Recommended Citation Liu, Chunyu, "Quinic Acid-Mediated Induction of Hypovirulence and a Hypovirulence-Associated Double-Stranded RNA in Rhizoctonia Solani" (2001). Electronic Theses and Dissertations. 332. http://digitalcommons.library.umaine.edu/etd/332 This Open-Access Dissertation is brought to you for free and open access by DigitalCommons@UMaine. It has been accepted for inclusion in Electronic Theses and Dissertations by an authorized administrator of DigitalCommons@UMaine. QUlNlC ACID-MEDIATED INDUCTION OF HYPOVIRULENCE AND A HY POVlRULENCE-ASSOCIATED DOUBLESTRANDED RNA IN RHIZOCTONIA SOLANI BY Chunyu Liu B.S. Wuhan University, 1989 MS. Wuhan University. 1992 A THESIS Submitted in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy (in Biochemistry and Molecular Biology) The Graduate School The Universrty of Maine August, 2001 Advisory Committee: Stellos Tavantzis, Professor of Plant Pathology, Advisor Seanna Annis, Assistant Professor of Mycology Robert Cashon, Assistant Professor of Biochemistry, Molecular Biology Robert Gundersen, Associate Professor of Biochemistry, Molecular Biology John Singer, Professor of Microbiology QUlNlC ACID-MEDIATED INDUCTION OF HYPOVIRULENCE AND A HYPOVIRULENCE-ASSOCIATED DOUBLE-STRANDED RNA (DSRNA) IN RHIZOCTONIA SOLANI By Chunyu Liu Thesis Advisor: Dr. Stellos Tavantzis An Abstract of the Thesis Presented in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy (in Biochemistry and Molecular Biology) August, 2001 This study is a part of a project focused on the relationship between dsRNA and hypovirulence in R. -

Downloaded As a Text File, Is Completely Dynamic
BMC Bioinformatics BioMed Central Database Open Access ORENZA: a web resource for studying ORphan ENZyme activities Olivier Lespinet and Bernard Labedan* Address: Institut de Génétique et Microbiologie, CNRS UMR 8621, Université Paris-Sud, Bâtiment 400, 91405 Orsay Cedex, France Email: Olivier Lespinet - [email protected]; Bernard Labedan* - [email protected] * Corresponding author Published: 06 October 2006 Received: 25 July 2006 Accepted: 06 October 2006 BMC Bioinformatics 2006, 7:436 doi:10.1186/1471-2105-7-436 This article is available from: http://www.biomedcentral.com/1471-2105/7/436 © 2006 Lespinet and Labedan; licensee BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Abstract Background: Despite the current availability of several hundreds of thousands of amino acid sequences, more than 36% of the enzyme activities (EC numbers) defined by the Nomenclature Committee of the International Union of Biochemistry and Molecular Biology (NC-IUBMB) are not associated with any amino acid sequence in major public databases. This wide gap separating knowledge of biochemical function and sequence information is found for nearly all classes of enzymes. Thus, there is an urgent need to explore these sequence-less EC numbers, in order to progressively close this gap. Description: We designed ORENZA, a PostgreSQL database of ORphan ENZyme Activities, to collate information about the EC numbers defined by the NC-IUBMB with specific emphasis on orphan enzyme activities. -
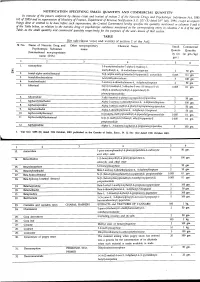
NOTIFICATION SPECIFYING SMALL QUANTITY and COMMERCIAL QUANTITYI Ly of the Pozoersconferred (Xxiiiil
NOTIFICATION SPECIFYING SMALL QUANTITY AND COMMERCIAL QUANTITYI ly of the pozoersconferred (xxiiiil . - lleyise by clansesfuiia) and of section2 of the Nnrcotic Drrtgsnnd pnlchotropic SrtbstancesAct, 19g5 (61 of 1985)and in supersessionof Ministry of Ftnnnce, (D Departmentof'Reuenue Noiification s.o.527 aotra i'6tt,1uli, tssi, exceptas respects things doneor omitted to be done .beforesuch supersession,the Cential Gooernmenihereby spectfiesthe qtnntitrl ,nlnt'ionedin coltnnnsS and 6 of the Tablebelow, in relationto.the narcoticdrug or psychotropicsubstnnce mentioned.ii tlr', ,1urrponding entnl in columns2 to 4 of thesaid Table,as the small quantitrl nnd commercialquantity respectiailyfor the purposesof the said clnuse:sof ttrit seciion. TABLE [Seesub-clause vii(a) and xxiii(a) of section 2 of the Act] Sl No. Name of Narcotic Drug and Other non-proprietary Chemical Name Small Commercial Psychotropic Substance name Quanti- Quantity (Intemational non-proprietary ity (in (in gm./kg.) name (INN) Acetorphine 3-0-acetyltetrahydro-7-alpha-(l-hydroxy-l- methylbutyt)-o, l4-endoetheno-onpavine $ 50 9.. 1\) Acetyl-alpha-methylfen N-[-(alpha-methylphenethyl)-4-piperidy]l acetanilide 0.1 g-. J. Acetyldihydrocodeine 100 4. Acetylmethadol 3-acetoxy-6-dimethylamino-4, 4 lheptane 50 gm. 5. Alfentanil -ethyl-4, N-[1-[2-( 5-dihydro-S-oxo-lH-tetrazol-t-yt) 01 gm. ethyll-4-(methoxymethyl)-4-piperidinyll -N- ylpropanamide Allyprodine 3-allyl Jmethyl-4-phenyl-4 Alpha-3-acetoxy-6-dimethylamino-4,Ld lheptane 100 gm Alpha-3-ethyl-l-methyl-4-phenyl-4-propionox 50 gm. 9.7' AlphamethadolArPnametnaool Alpha-6-dimethylamino-4, 4-diphenyl-3-heptanol 2 S0 gm. 10. Alphu-*"thylf"ntur,yl 11. -

0 Cover Part Two\374
PART TWO DEUXIÈME PARTIE SEGUNDA PARTE ﺍﳉﺰﺀ ﺍﻟﺜﺎﱐ 第二部分 ЧАСТЬ ВТΟΡАЯ - (...), [...] - (-)-3-hidroxi-N- (-)-(2R)-N-méthyl-1- fenacilmorfinán → Levophenacyl-morphan phénylpropan-2-amine → Levometamfetamine (-)-3-hidroxi-N- (-)-(3S,6R)-6- metilmorfinán → Levorphanol (dimethylamino)-4,4- (-)-3-hydroksymorfinan → Norlevorphanol diphenyl-3-heptanol → Betamethadol (-)-3-hydroksy-N- (-)-(3S,6R)-6- fenacylmorfinan → Levophenacyl-morphan (dimethylamino)-4,4- (-)-3-hydroksy-N- diphenyl-3-heptanol metylmorfinan → Levorphanol acetate (ester) → Betacetylmethadol (-)-3-hydroxymorphinan → Norlevorphanol (-)-(5R)-4,5-epoxy-3- (-)-3-hydroxy-N- methoxy-9α-methyl methylmorphinan → Levorphanol → Thebacon morphin-6-en-6-yl acetate (-)-3-hydroxynormorphinan → Norlevorphanol (-)-(5R,6S)-3-benzyloxy-4,5- (-)-3-hydroxy-N- epoxy-9a-methylmorphin- phenacylmorphinan → Levophenacyl-morphan 7-en-6-yl myristate → Myrophine (-)-3-hydroxytropane-2- (-)-(5R,6S)-4,5-epoxy carboxylat → Ecgonine morphin-7-en-3,6-diol → Normorphine (-)-3-idrossi-N- (-)-(5R,6S,14S)-4,5-epoxy- metilmorfinano → Levorphanol 14-hydroxy-3-methoxy- (-)-3-methoxy-N- 9a-methylmorphinan-6- methylmorphinan → Levomethorphan one → Oxycodone (-)-3-metil-2,2-difenil-4- (-)-(R)-4,5-epoxy-3- morfolinbutirilpirrolidina → Levomoramide methoxy-9a-methyl (-)-3-metoksy-N- morphinan-6-one → Hydrocodone metylmorfinan → Levomethorphan (-)-(R)-6-(dimethylamino)- (-)-3-metossi-N-metil- 4,4-diphenyl-3-heptanone → l-methadone morfinano → Levomethorphan (-)-(R)-N,α-dimethyl (-)-3-metoxi-N- phenethylamine → Levometamfetamine -

Proof of De Novo Synthesis of the Qa Enzymes of Neurospora Crassa During Induction
Proc. Nati. Acad. Sci. USA Vol. 74, No. 10, pp. 4256-4260, October 1977 Biochemistry Proof of de novo synthesis of the qa enzymes of Neurospora crassa during induction [catabolic dehydroquinase (5-dehydroquinate dehydratase)/quinate dehydrogenase/qa gene cluster/eukaryote gene regulation] WILLIAM R. REINERT AND NORMAN H. GILES Program in Genetics, Department of Zoology, University of Georgia, Athens, Georgia 30602 Contributed by Norman H. Giles, July 22, 1977 ABSTRACT In Neurospora crassa three inducible enzymes There is strong genetic evidence that the qa-1 gene encodes a are necessary to catabolize quinic acid to protocatechuic acid. regulatory protein which, in the presence of quinic acid, exerts The three genes encoding these enzymes are tightly linked on chromosome VII near methionine-7 (me-7). This qa cluster in- positive control over the synthesis of the enzymes encoded in cludes a fourth gene, qa-1, which encodes a regulatory protein the three adjacent structural genes (6, 10). The appearance of apparently exerting positive control over transcription of the enzyme activity is very specifically regulated. In the presence other three qa genes. However, an alternative hypothesis is that of a preferred carbon source, e.g., sucrose, the activities of all the qa-I protein simply activates preformed polypeptides de- three enzymes are quite low. Quinate as a sole carbon source rived from the three structural genes. The use of density labeling causes a coordinate induction of all three enzymes (9). This with D20 demonstrated conclusively that the qa enzymes are synthesized de novo only during induction on quinic acid. Na- increase in enzyme activity can be inhibited by the simulta- tive catabolic dehydroquinase (5-dehydroquinate dehydratase; neous addition of cycloheximide to the medium (ref. -

Ep 2398808 B1
(19) TZZ ¥Z_T (11) EP 2 398 808 B1 (12) EUROPEAN PATENT SPECIFICATION (45) Date of publication and mention (51) Int Cl.: of the grant of the patent: C07D 489/08 (2006.01) 20.11.2013 Bulletin 2013/47 (86) International application number: (21) Application number: 10704301.0 PCT/US2010/024372 (22) Date of filing: 17.02.2010 (87) International publication number: WO 2010/096416 (26.08.2010 Gazette 2010/34) (54) Process for the reductive alkylation of normorphinans VERFAHREN ZUR REDUKTIVEN ALKYLIERUNG VON NORMORPHINANEN Procédé d’alkylation réductrice des normorphinanes (84) Designated Contracting States: • WOODS, Sharon AT BE BG CH CY CZ DE DK EE ES FI FR GB GR Florissant HR HU IE IS IT LI LT LU LV MC MK MT NL NO PL Missouri 63034 (US) PT RO SE SI SK SM TR (74) Representative: Beckmann, Claus (30) Priority: 17.02.2009 US 153021 P Kraus & Weisert Patent- und Rechtsanwälte (43) Date of publication of application: Thomas-Wimmer-Ring 15 28.12.2011 Bulletin 2011/52 80539 München (DE) (73) Proprietor: Mallinckrodt LLC (56) References cited: Hazelwood, MO 63042 (US) WO-A1-2006/035195 WO-A1-2009/012005 (72) Inventors: • HUDSON, Edmund, C. Clayton Missouri 63105 (US) Note: Within nine months of the publication of the mention of the grant of the European patent in the European Patent Bulletin, any person may give notice to the European Patent Office of opposition to that patent, in accordance with the Implementing Regulations. Notice of opposition shall not be deemed to have been filed until the opposition fee has been paid. -

Back Matter (PDF)
The Editor of the Journal of Pharmacology & Experimental Therapeutics wishes to express appreciation to the following colleagues who acted as Guest Editors for Specific Fields in 1987. Martin W. Adler Frank R. Goodman Elaine Sanders-Bush Lewis Aronow Robert Z. Gussin Lewis E. Seiden C. Paul Bianchi Conan Kornetsky Theodore J. Torphy Allan H. Conney Donald J. Jenden John L. Skosey Theodore J. Cicero Robert M. Joy Larry G. Stark Charles R. Craig Louis Lemberger Norman Weiner Gerard L. Gebber Benedict R. Lucchesi Wallace D. Winters James W. Gibb Donald V. Priola Dixon M. Woodbury Dora B. Goldstein Additionally, sincere thanks are due to 724 of our colleagues not on the Editorial Advisory Board who reviewed articles for the Journal in 1987. Their contributions are acknowledged individually in the Annual Report of The Journal to The Board of Publications Trustees of The American Society for Pharmacology and Experimental Therapeutics. INDEX Volume 245, April-June, 1988 A23187, effect ofextracellularcalcium omis- classification, ligand affmity and mo- -induced intoxication, effects of Ro15- sion, hepatocytes (rats), 493 lecular mass (bovine), 1060 4513 (rats), 880 Abhold, R. H. and Harding, J. W.: Metabo- imidazoline analogs, aorta (rats), 793 Mi, S. F., see Schulze, G. E., 178 lism of angiotensins II and III by inotropic positive effect, inositol tris- Alkondon, M., Rao, K. S. and Albuquerque, membrane-bound peptidases from phosphate increase in heart (rats), E. X.: Acetylcholinesterase reactiva- rat brain, 171 327 tom modify the functional properties Abiko, Y., see Hara, Y., 305 prazosin and indoramin, coronary of the nicotinic acetylcholine recep- Acetaminophen blood flow (dogs), 232 tor ion channel, 543 diminuition of hepatotoxicity, ethanol alpha-2 Allbee, W. -

Subcellular Localization of Quinate: Oxidoreductase from Phaseolus Mungo L. Sprouts Xiangbo Kang, H
Subcellular Localization of Quinate: Oxidoreductase from Phaseolus mungo L. Sprouts Xiangbo Kang, H. Ekkehard Neuhaus and Renate Scheibe Pflanzenphysiologie, Fachbereich Biologie/Chemie, Universität Osnabrück, D-49069 Osnabrück, Bundesrepublik Deutschland Z. Naturforsch. 49c, 415-420 (1994); received March 24/April 27, 1994 Phaseolus mungo, Quinate:Oxidoreductase, Quinic Acid, Prechorismate Pathway, Shikimate Pathway Quinate:oxidoreductase (QORase, EC 1.1.1.24) was isolated and purified from etiolated mung bean (Phaseolus mungo L.) sprouts and a monospecific antiserum was raised in rabbit to the homogeneous protein. Highly intact etioplasts were isolated from the same plant material. The stroma of the purified etioplasts was enzymatically characterized. Contami nation by cytosol, mitochondria and vacuole was estimated from activities of marker en zymes. QORase activity was localized in the stroma (about 91% for both NAD+ and NADP+ as a cofactor). Western blotting and immunoprinting of the stroma proteins revealed a single band that migrated identically with the purified QORase. The results suggest that the QOR ase is localized predominantly, if not exclusively, in the etioplast stroma. The physiological role of the enzyme is discussed. Introduction (Refeno et al., 1982), tobacco callus (Beaudoin Quinic acid is widely distributed among higher and Thorpe, 1983, 1984) and conifer needles (Osi and lower plants (Karrer, 1958; Minamikawa and pov and Shein, 1987). Two of them were reported Yoshida, 1972; Boudet, 1973; Yoshida et al, 1975). to be purified to homogeneity (Refeno et al., 1982; Therefore, its physiological function and metab Kang and Scheibe, 1993). However, the character olism are of interest. At the turn of the 1960’s istics of these enzymes appeared to be very diver Weinstein et al. -

Recent Advances in the Chemistry of Oripavine and Its Derivatives
Advances in Bioscience and Biotechnology, 2014, 5, 704-717 Published Online July 2014 in SciRes. http://www.scirp.org/journal/abb http://dx.doi.org/10.4236/abb.2014.58084 Recent Advances in the Chemistry of Oripavine and Its Derivatives Sandor Hosztafi Institute of Pharmaceutical Chemistry, Semmelweis University, Budapest, Hungary Email: [email protected] Received 2 May 2014; revised 16 June 2014; accepted 12 July 2014 Copyright © 2014 by author and Scientific Research Publishing Inc. This work is licensed under the Creative Commons Attribution International License (CC BY). http://creativecommons.org/licenses/by/4.0/ Abstract Oripavine is the major alkaloid of Papaver orientale. It is an important intermediate in the bio- synthesis of morphine alkaloids. Recently, new Papaver somniferum strains have been developed which accumulate thebaine and oripavine, but not morphine and codeine. Therefore, the chemi- stry of oripavine has been studied intensively to synthesize opioid pharmaceuticals such as oxy- morphone, naloxone and buprenorphine. Keywords Papaver orientale, Papaver somniferum, Oripavine, Thebaine, Top1 Poppy 1. Introduction Thebaine an alkaloid present in 0.2% - 0.8% in opium and a major constituent (90% of total alkaloid content) in Papaver bracteatum (which is morphine free), possesses little utility medically for two reasons: (a) its lack of the depressant and analgesic properties common to other morphine alkaloids and (b) its expression of extreme toxicity and CNS stimulation. Oripavine is also a minor alkaloid of the opium poppy, but it is the main alkaloid of the oriental poppy Papaver orientale, which is a perennial flowering plant. The development of the top1 poppy was an important finding for the opium industry in Australia, because this poppy strain produces thebaine and oripavine, but not morphine or codeine. -

All Enzymes in BRENDA™ the Comprehensive Enzyme Information System
All enzymes in BRENDA™ The Comprehensive Enzyme Information System http://www.brenda-enzymes.org/index.php4?page=information/all_enzymes.php4 1.1.1.1 alcohol dehydrogenase 1.1.1.B1 D-arabitol-phosphate dehydrogenase 1.1.1.2 alcohol dehydrogenase (NADP+) 1.1.1.B3 (S)-specific secondary alcohol dehydrogenase 1.1.1.3 homoserine dehydrogenase 1.1.1.B4 (R)-specific secondary alcohol dehydrogenase 1.1.1.4 (R,R)-butanediol dehydrogenase 1.1.1.5 acetoin dehydrogenase 1.1.1.B5 NADP-retinol dehydrogenase 1.1.1.6 glycerol dehydrogenase 1.1.1.7 propanediol-phosphate dehydrogenase 1.1.1.8 glycerol-3-phosphate dehydrogenase (NAD+) 1.1.1.9 D-xylulose reductase 1.1.1.10 L-xylulose reductase 1.1.1.11 D-arabinitol 4-dehydrogenase 1.1.1.12 L-arabinitol 4-dehydrogenase 1.1.1.13 L-arabinitol 2-dehydrogenase 1.1.1.14 L-iditol 2-dehydrogenase 1.1.1.15 D-iditol 2-dehydrogenase 1.1.1.16 galactitol 2-dehydrogenase 1.1.1.17 mannitol-1-phosphate 5-dehydrogenase 1.1.1.18 inositol 2-dehydrogenase 1.1.1.19 glucuronate reductase 1.1.1.20 glucuronolactone reductase 1.1.1.21 aldehyde reductase 1.1.1.22 UDP-glucose 6-dehydrogenase 1.1.1.23 histidinol dehydrogenase 1.1.1.24 quinate dehydrogenase 1.1.1.25 shikimate dehydrogenase 1.1.1.26 glyoxylate reductase 1.1.1.27 L-lactate dehydrogenase 1.1.1.28 D-lactate dehydrogenase 1.1.1.29 glycerate dehydrogenase 1.1.1.30 3-hydroxybutyrate dehydrogenase 1.1.1.31 3-hydroxyisobutyrate dehydrogenase 1.1.1.32 mevaldate reductase 1.1.1.33 mevaldate reductase (NADPH) 1.1.1.34 hydroxymethylglutaryl-CoA reductase (NADPH) 1.1.1.35 3-hydroxyacyl-CoA