Morphinone Reductase for the Preparation Of
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

In the United States District Court for the Eastern District of Pennsylvania ______: in Re Suboxone (Buprenorphine : Mdl No
IN THE UNITED STATES DISTRICT COURT FOR THE EASTERN DISTRICT OF PENNSYLVANIA __________________________________________ : IN RE SUBOXONE (BUPRENORPHINE : MDL NO. 2445 HYDROCHLORIDE AND NALOXONE) : 13-MD-2445 ANTITRUST LITIGATION : : THIS DOCUMENT RELATES TO:, : : Wisconsin, et al. v. Indivior Inc. et al. : Case No. 16-cv-5073 : __________________________________________: STATE OF WISCONSIN : By Attorney General Brad D. Schimel, et al. : : CIV. A. NO. 16-5073 Plaintiffs, : v. : : INDIVIOR INC. f/k/a RECKITT BENCKISER : PHARMACEUTICALS, INC., et al. : : Defendants. : __________________________________________: Goldberg, J. November 24, 2020 MEMORANDUM Defendant Reckitt Benckiser, Inc. (“Defendant”) manufactures Suboxone, a drug commonly used to combat opioid addiction.1 Suboxone previously came in tablet form, but in 2010, citing safety concerns, Defendant effectuated a change in the administration of this drug, switching from tablet to sublingual film. Various purchasers/consumers of Suboxone claimed that this switch was anticompetitive and solely designed to maintain Defendant’s market exclusivity—a scheme known as a “product hop.” These claims have resulted in multi-district, antitrust litigation before this Court. 1 Reckitt is currently known as Indivior, Inc. In December 2014, Reckitt Benckiser Pharmaceuticals, Inc. was demerged from its prior parent, the Reckitt Benckiser Group PLC, into Indivior PLC. Although Indivior is technically the named defendant in this case, the pleadings and many of the relevant exhibits use the name “Reckitt.” As discovery and class certification litigation have come to a close, the parties have raised numerous challenges under Daubert v. Merrell Dow Pharmaceuticals, 509 U.S. 579 (1993), seeking exclusion of all or selected portions of nine expert witnesses anticipated opinions. This Opinion explains my reasoning for the resolution of these motions and will hopefully set forth a clearer path towards trial. -

Alternative Formats If You Require This Document in an Alternative Format, Please Contact: [email protected]
University of Bath PHD The extraction and chemistry of the metabolites of Mimosa tenuiflora and Papaver somniferum Ninan, Aleyamma Award date: 1990 Awarding institution: University of Bath Link to publication Alternative formats If you require this document in an alternative format, please contact: [email protected] General rights Copyright and moral rights for the publications made accessible in the public portal are retained by the authors and/or other copyright owners and it is a condition of accessing publications that users recognise and abide by the legal requirements associated with these rights. • Users may download and print one copy of any publication from the public portal for the purpose of private study or research. • You may not further distribute the material or use it for any profit-making activity or commercial gain • You may freely distribute the URL identifying the publication in the public portal ? Take down policy If you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediately and investigate your claim. Download date: 23. Sep. 2021 THE EXTRACTION AND CHEMISTRY OF THE METABOLITES OF MIMOSA TENUIFLORA AND PAP AVER SOMNIFERUM. submitted by ALEYAMMA NINAN for the degree of Doctor of Philosophy of the University of Bath 1990 Attention is drawn to the fact that the copyright of this thesis rests with its author. This copy of the thesis has been supplied on condition that anyone who consults it is understood to recognise that its copyright rests with its author and that no quotation from the thesis and no information derived from it may be published without prior consent of the author. -

The Metamorphosis of Hydromorphone Gary M
Pharmacy PersPective The metamorphosis of hydromorphone Gary M. Reisfield, MD George R. Wilson, MD introduction “That’s it. The Mayo Clinic at Rochester devel - oped it, the word and the drug, for it means a Hydromorphone hydrochloride, one of the oldest of drug, a pain relieving drug, five times as potent the extant opioid analgesics, has been in clinical use for as morphine, as harmless as water and with no more than 70 years. Its use by the oral route in chronic habit forming qualities. pain and hospice/palliative medicine settings has been limited, however, largely owing to its relatively short “The medical journals say it is particularly useful duration of action. With the recent US Food and Drug in the operation of cases where other drugs Administration (FDA) approval of a once-daily extended- seem to offer no relief from pain. Unlike mor - release formulation of the drug (Palladone, Purdue phine, there are no pleasurable sensations to its Pharma LP, Stamford, CT), hydromorphone joins mor - use, however, and if the doctors reckon correctly phine, oxycodone, and fentanyl as the only extended- its use may go far toward curing addicts of the release opioids available on the United States market. morphine habit.” Here, we review the history, pharmacokinetics, and other relevant issues concerning this invaluable opioid, and Montgomery (AL) Advertiser , Dec. 18, 1932 also discuss the role of the new formulation in the man - agement of chronic pain. From 1929 to 1939, the National Research Council’s Committee on Drug Addiction conducted exhaustive history research on the morphine molecule and its analogs, pro - ducing more than 150 semisynthetic and more than 300 Hydromorphone [also Dilaudid (Knoll Laboratories, synthetic compounds, of which more than 30 were tested Mount Olive, NJ), dihydromorphinone, dihydromorphe - clinically. -

FREDERICK ('ERIC') RANDALL SMITH Bsc, Phd(Edin), FRSC Eric
FREDERICK ('ERIC') RANDALL SMITH BSc, PhD(Edin), FRSC Eric Smith was responsible for research activities in Duncan Flockhart Ltd, T & H Smith Ltd and Edinburgh Pharmaceutical Industries Ltd during the period 1947 to 1971. He and his colleagues made several lasting contributions to medicine and commerce notable amongst which were the creation of a new opiate industry based on large scale poppy growing in Australia, the establishment of dihydrocodeine ('DF118') as a major analgesic, and the discovery of 'Bitrex', an intensely bitter substance, now a widely used denaturant. He was elected a Fellow of the Society in 1962 and served on Council 1969-71. Born in Edinburgh on April 4th, 1911, Eric spent his childhood, his school and university days, and most of his working life and retirement in or near to the City. He died there on November 4th, 1994. Eric's enquiring attitude to life owed much to his father. This unusual man earned his living in the Actuarial Department of the Scottish Provident Institution but his main interests were astronomy and art. He was an FRAS and his personal observations led to his name being given to a minor lunar crater, and a good enough artist for his works to be hung in the Royal Academies in Edinburgh and London. Perhaps this is why Eric chose to be a scientist and his sister an art teacher. Eric was educated at the Edinburgh Institution (later named Melville College) where he was an excellent scholar and a good enough sportsman to play for the 1st XV and the FPs. -

PHARMACEUTICALS + Biovian Oy Jemedic AB Bayer Oy + Shetland Bergen Helse Bergen HF, Haukeland 3
BARENTS SEA Jan Mayen (Norway) Tromsø NORWAY Murmansk DENMARK STRAIT NORWEGIAN SEA Raufarhöfn Bolungarvik Kalix Aromtech Oy Mosjøen SWEDEN Kemi Akureyri Luleå WHITE SEA Piteå Eskifjördur Oy Woikoski Ab + Pharmatory Ltd + Bioactive Bone Substitutes Oolu Fermion Oy Olafsvik ICELAND Skellefteå IA Hornafjördur Reykjavik 1-5 Grindavik Apotek Produktion & Laboratorier AB Oy Woikoski Ab Umeå Norrlands Universitetssjukhus Frøya Vik Trondheim Kristiansund GULF OF BOTHN Vaasa Averøya Galena Pharma Oy + Orion Oyj + Finvector Oy Sandøy Härnösand Ålesund Sundsvall Unimedic AB + MAP Medical Technologies Oy Woikoski Ab Kristinestad Tórshavn Apoteksverkiö, Framleiösluapotekiö Måløy EWOS AS Santen Oy Cytomed Oy SWEDEN - Not Shown Vitabalans Oy + Umeå + Curida AS Apotek Produktion & Laboratorier AB Oy Aga Ab Norrlands Universitetssjukhus Jemedic AB FINLAND OSLO Gävle Alby Vyborg + Nouryon Pulp & Performance Chemicals AB Hankintatukku Oy 1. Smerud Medical Research Norway AS Pcas Finland Oy Finex Oy Hamina Orion Corporation Oy Woikoski Ab 2. Catapult Life Science AS Gävle + + EUROPE - PHARMACEUTICALS + Biovian Oy Jemedic AB Bayer Oy + Shetland Bergen Helse Bergen HF, Haukeland 3. PhotoCure ASA Turku Orion Corporation A.Vogel Oy universitetssjukehus 4. GE Healthcare AS Vitabalans Oy MAP Medicals Technologies Oy + Nanoform Finland Oy Islands 5. Oncoinvent AS Fermion Oy AGA Gas AB Lumene Oy Lerwick 6. The Cyclotron Center Pharmaq AS + Scan Aqua AS Orion Corporation + 7. Oslo Universitetssykehus HF HELSINKI 8. Diatec Monoclonals AS Institute for Energy Technology -

Effect of Anticancer Agents on Codeinone-Induced Apoptosis in Human Cancer Cell Lines
ANTICANCER RESEARCH 25: 4037-4042 (2005) Effect of Anticancer Agents on Codeinone-induced Apoptosis in Human Cancer Cell Lines RISA TAKEUCHI1,2, HIROSHI HOSHIJIMA1,2, NORIKO ONUKI1,2, HIROSHI NAGASAKA1, SHAHEAD ALI CHOWDHURY3, MASAMI KAWASE4 and HIROSHI SAKAGAMI2 1Division of Anesthesiology, Department of Comprehensive Medical Sciences, 2Division of Pharmacology, Department of Diagnostic and Therapeutic Sciences, and 3MPL (Meikai Phamaco-Medical Laboratory), Meikai University School of Dentistry, Sakado, Saitama 350-0283; 4Faculty of Phamaceutical Sciences, Josai University, Sakado, Saitama 350-0295, Japan Abstract. The possible apoptosis-inducing activity of Opioids have been given to patients with cancer pain, codeinone, an oxidative metabolite of codeine, without or according to the WHO ladder against pain (4). According with anticancer drugs, was investigated. Codeinone induced to reports of Ventafridda et al., pain occurs in more than internucleosomal DNA fragmentation in human 50% of cancer patients, and the administration of opioids promyelocytic leukemia cells (HL-60), but not in human manifests significantly higher therapeutic effects against squamous cell carcinoma cells (HSC-2). Codeinone dose- cancer pain, compared with non-narcotics (5, 6). Although dependently activated caspase-3 in both of these cells, but to opioids have been used for patients in clinical situations, the a much lesser extent than that attained by actinomycin D. anticancer action of opioids has not been well investigated. This property of codeinone was similar to what we have Both opioids and anticancer drugs have been simultaneously found previously in ·,‚-unsaturated ketones. Codeinone did given for pain in cancer patients. However, there has been not activate caspase-8 or caspase-9 in these cells. -

Microbial Degradation of the Morphine Alkaloids Purification and Characterization of Morphine Dehydrogenase from Pseudomonas Putida M10
Biochem. J. (1991) 274, 875-880 (Printed in Great Britain) 875 Microbial degradation of the morphine alkaloids Purification and characterization of morphine dehydrogenase from Pseudomonas putida M10 Neil C. BRUCE,* Clare J. WILMOT, Keith N. JORDAN, Lauren D. Gray STEPHENS and Christopher R. LOWE Institute of Biotechnology, University of Cambridge, Tennis Court Road, Cambridge CB2 1QT, U.K. The NADP+-dependent morphine dehydrogenase that catalyses the oxidation of morphine to morphinone was detected in glucose-grown cells of Pseudomonas putida M 10. A rapid and reliable purification procedure involving two consecutive affinity chromatography steps on immobilized dyes was developed for purifying the enzyme 1216-fold to electrophoretic homogeneity from P. putida M 10. Morphine dehydrogenase was found to be a monomer of Mr 32000 and highly specific with regard to substrates, oxidizing only the C-6 hydroxy group of morphine and codeine. The pH optimum of morphine dehydrogenase was 9.5, and at pH 6.5 in the presence of NADPH the enzyme catalyses the reduction of codeinone to codeine. The Km values for morphine and codeine were 0.46 mm and 0.044 mm respectively. The enzyme was inhibited by thiol-blocking reagents and the metal-complexing reagents 1, 10-phenanthroline and 2,2'-dipyridyl, suggesting that a metal centre may be necessary for activity of the enzyme. INTRODUCTION EXPERIMENTAL The morphine alkaloids have attracted considerable attention Materials owing to their analgaesic properties and, consequently, much Mimetic Orange 3 A6XL and Mimetic Red A6XL were effort in the past has been directed at the production of new obtained from Affinity Chromatography Ltd., Freeport, morphine alkaloids by micro-organisms (lizuka et al., 1960, Ballasalla, Isle of Man, U.K. -

United States Securities and Exchange Commission Form 10-K Shire Pharmaceuticals Group
UNITED STATES SECURITIES AND EXCHANGE COMMISSION WASHINGTON, D.C. 20549 FORM 10-K (Mark One) ፤ ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 For the fiscal year ended December 31, 1999 អ TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 Commission file number 0-29630 SHIRE PHARMACEUTICALS GROUP PLC (Exact name of registrant as specified in its charter) England and Wales (State or other jurisdiction (I.R.S. Employer of incorporation or organization) Identification No.) N.A. East Anton, Andover, Hampshire SP10 5RG England (Address of principal executive offices) (Zip Code) 44 1264 333455 (Registrant's telephone number, including area code) Securities registered pursuant to Section 12(b) of the Act: Title of each class Name of exchange on which registered American Depository Shares, each representing Nasdaq National Market 3 Ordinary Shares, 5 pence nominal value per share Securities registered pursuant to Section 12(g) of the Act: None (Title of class) Indicate by check mark whether the Registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the Registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ፤ No អ Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of the Registrant's knowledge, in definitive proxy or information statements incorporated by reference to Part III of this Form 10-K or any amendment to this Form 10-K. -

August/September 2016
INTERNATIONAL PHARMACEUTICAL QUALITY IPQ Inside the Global Regulatory Dialogue IPQPUBS.COM VOL. 8, NO. 5 MONTHLY UPDATE - AUGUST/SEPTEMBER 2016 UNITED STATES ● Review/Inspection Control Strategy Communications Key to Advancing Biotech p. 4 Regulation, FDA Biotech Official Stresses ● More Clarity Sought in FDA’s Inactive Ingredient Database Regarding Drug Delivery Devices p. 13 EUROPE ● Protein Sequence Variants, Process Development and Spec Setting Are Generating Queries p. 20 from EU Biopharmaceutical CMC Assessors ● Procedures, Eligibilities and Goals of EMA’s Accelerated Access Efforts Are Taking More p. 32 Concrete Shape INTERNATIONAL ● Goal of FDA/EU Mutual Inspection Reliance is Nearing Realization p. 36 UPDATES IN BRIEF p. 47 U.S. CMC: ● GDUFA II Agreement ● ANDA Impurity Refusals ● Capsule Supplier Changes ● Co-Crystal Classification ● ANDA Impurity Refusals U.S. GMP: ● Insanitary Compounding ● NIH Clinical Manufacturing ● Establishment Registration ● Generics Facility ID'ing ● USP on Analytical Lifecycles EUROPE CMC: ● EMA Streamlining ● EMA Adaptive Pathways Workshop ● EDQM on ICH Q3D EUROPE GMP: ● EMA WFI Q&A ● EC on Plastics INTERNATIONAL CMC: ● Antimicrobial Resistance Roadmap ● WHO on Variations ● Canada on ICH Q3D INTERNATIONAL GMP: ● China on Drug Manufacturing ● India Manufacturing Self-Assessment FDA WARNING LETTERS, EMA NON-COMPLIANCE REPORTS, AND FDA RECALLS POSTED IN AUGUST/SEPTEMBER p. 51 MONTHLY UPDATE - AUGUST / SEPTEMBER 2016 INTERNATIONAL PHARMACEUTICAL QUALITY provides in-depth coverage of emerging drug, biologic and combination product CMC and GMP issues and developments with a mission of helping advance and harmonize the quality regulatory process globally. Headquartered in Washington, D.C., IPQ is read by regulatory agencies, manufacturers, suppliers, consultants, law firms, and universities around the world. -

United States Patent Office Patented Aug
3,830,819 United States Patent Office Patented Aug. 20, 1974 2 The invention provides a process for the purification of l-dihydrocodeine containing as impurities 1-dihydrothebai 3,830,819 MANUFACTURE OF l-DHYDROCODENE none and or 1-dihydrothebainol, which comprises treating Edward Leon Grew and David Jackson Powles, Edin the impure 1-dihydrocodeine to remove said impurities burgh, Scotland, assignors to MacFarlan Smith Limited, therefrom by either extraction with an aqueous alkali or Edinburgh, Scotland 5 by passage of the material through an anion exchange No Drawing. Fied Feb. 22, 1972, Ser. No. 228,367 resin of a quaternary ammonium type in the hydroxide Claims priority, application Great Britain, Feb. 22, 1971, form whereby the proportion of said impurities is reduced. 5,081/71 The crude 1-dihydrocodeine may be one prepared by Int, C. C07d 43/28 the catalytic hydrogenation of 1-dihydrocodeinone and ac U.S. C. 260-285 3 Claims 0. cording to a feature of the invention, there is therefore provided a process for the preparation of a purified 1-di ABSTRACT OF THE DESCLOSURE hydrocodeine which comprises the steps of A process for the preparation of a purified 1-dihydro (a) catalytically hydrogenating 1-dihydrocodeinone in a codeine which comprises the steps of: 5 liquid medium using a platinum oxide or supported (a) catalytically hydrogenating 1-dihydrocodeinone in a platinum metal catalyst to produce a solution of crude liquid medium using a catalyst selected from the group l-dihydrocodeine, consisting of platinum oxide and supported platinum (b) removing the catalyst from the solution, and metal to produce a solution of crude 1-dihydrocodeine, (c) treating the solution for the recovery of a purified (b) removing the catalyst from the solution, and 20 1-dihydrocodeine having a reduced content of l-dihy (c) treating the solution for recovery of a purified 1-di drothebainone and/or 1-dihydrothebainol. -
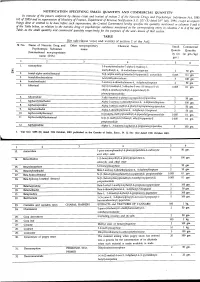
NOTIFICATION SPECIFYING SMALL QUANTITY and COMMERCIAL QUANTITYI Ly of the Pozoersconferred (Xxiiiil
NOTIFICATION SPECIFYING SMALL QUANTITY AND COMMERCIAL QUANTITYI ly of the pozoersconferred (xxiiiil . - lleyise by clansesfuiia) and of section2 of the Nnrcotic Drrtgsnnd pnlchotropic SrtbstancesAct, 19g5 (61 of 1985)and in supersessionof Ministry of Ftnnnce, (D Departmentof'Reuenue Noiification s.o.527 aotra i'6tt,1uli, tssi, exceptas respects things doneor omitted to be done .beforesuch supersession,the Cential Gooernmenihereby spectfiesthe qtnntitrl ,nlnt'ionedin coltnnnsS and 6 of the Tablebelow, in relationto.the narcoticdrug or psychotropicsubstnnce mentioned.ii tlr', ,1urrponding entnl in columns2 to 4 of thesaid Table,as the small quantitrl nnd commercialquantity respectiailyfor the purposesof the said clnuse:sof ttrit seciion. TABLE [Seesub-clause vii(a) and xxiii(a) of section 2 of the Act] Sl No. Name of Narcotic Drug and Other non-proprietary Chemical Name Small Commercial Psychotropic Substance name Quanti- Quantity (Intemational non-proprietary ity (in (in gm./kg.) name (INN) Acetorphine 3-0-acetyltetrahydro-7-alpha-(l-hydroxy-l- methylbutyt)-o, l4-endoetheno-onpavine $ 50 9.. 1\) Acetyl-alpha-methylfen N-[-(alpha-methylphenethyl)-4-piperidy]l acetanilide 0.1 g-. J. Acetyldihydrocodeine 100 4. Acetylmethadol 3-acetoxy-6-dimethylamino-4, 4 lheptane 50 gm. 5. Alfentanil -ethyl-4, N-[1-[2-( 5-dihydro-S-oxo-lH-tetrazol-t-yt) 01 gm. ethyll-4-(methoxymethyl)-4-piperidinyll -N- ylpropanamide Allyprodine 3-allyl Jmethyl-4-phenyl-4 Alpha-3-acetoxy-6-dimethylamino-4,Ld lheptane 100 gm Alpha-3-ethyl-l-methyl-4-phenyl-4-propionox 50 gm. 9.7' AlphamethadolArPnametnaool Alpha-6-dimethylamino-4, 4-diphenyl-3-heptanol 2 S0 gm. 10. Alphu-*"thylf"ntur,yl 11. -

0 Cover Part Two\374
PART TWO DEUXIÈME PARTIE SEGUNDA PARTE ﺍﳉﺰﺀ ﺍﻟﺜﺎﱐ 第二部分 ЧАСТЬ ВТΟΡАЯ - (...), [...] - (-)-3-hidroxi-N- (-)-(2R)-N-méthyl-1- fenacilmorfinán → Levophenacyl-morphan phénylpropan-2-amine → Levometamfetamine (-)-3-hidroxi-N- (-)-(3S,6R)-6- metilmorfinán → Levorphanol (dimethylamino)-4,4- (-)-3-hydroksymorfinan → Norlevorphanol diphenyl-3-heptanol → Betamethadol (-)-3-hydroksy-N- (-)-(3S,6R)-6- fenacylmorfinan → Levophenacyl-morphan (dimethylamino)-4,4- (-)-3-hydroksy-N- diphenyl-3-heptanol metylmorfinan → Levorphanol acetate (ester) → Betacetylmethadol (-)-3-hydroxymorphinan → Norlevorphanol (-)-(5R)-4,5-epoxy-3- (-)-3-hydroxy-N- methoxy-9α-methyl methylmorphinan → Levorphanol → Thebacon morphin-6-en-6-yl acetate (-)-3-hydroxynormorphinan → Norlevorphanol (-)-(5R,6S)-3-benzyloxy-4,5- (-)-3-hydroxy-N- epoxy-9a-methylmorphin- phenacylmorphinan → Levophenacyl-morphan 7-en-6-yl myristate → Myrophine (-)-3-hydroxytropane-2- (-)-(5R,6S)-4,5-epoxy carboxylat → Ecgonine morphin-7-en-3,6-diol → Normorphine (-)-3-idrossi-N- (-)-(5R,6S,14S)-4,5-epoxy- metilmorfinano → Levorphanol 14-hydroxy-3-methoxy- (-)-3-methoxy-N- 9a-methylmorphinan-6- methylmorphinan → Levomethorphan one → Oxycodone (-)-3-metil-2,2-difenil-4- (-)-(R)-4,5-epoxy-3- morfolinbutirilpirrolidina → Levomoramide methoxy-9a-methyl (-)-3-metoksy-N- morphinan-6-one → Hydrocodone metylmorfinan → Levomethorphan (-)-(R)-6-(dimethylamino)- (-)-3-metossi-N-metil- 4,4-diphenyl-3-heptanone → l-methadone morfinano → Levomethorphan (-)-(R)-N,α-dimethyl (-)-3-metoxi-N- phenethylamine → Levometamfetamine