August/September 2016
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

In the United States District Court for the Eastern District of Pennsylvania ______: in Re Suboxone (Buprenorphine : Mdl No
IN THE UNITED STATES DISTRICT COURT FOR THE EASTERN DISTRICT OF PENNSYLVANIA __________________________________________ : IN RE SUBOXONE (BUPRENORPHINE : MDL NO. 2445 HYDROCHLORIDE AND NALOXONE) : 13-MD-2445 ANTITRUST LITIGATION : : THIS DOCUMENT RELATES TO:, : : Wisconsin, et al. v. Indivior Inc. et al. : Case No. 16-cv-5073 : __________________________________________: STATE OF WISCONSIN : By Attorney General Brad D. Schimel, et al. : : CIV. A. NO. 16-5073 Plaintiffs, : v. : : INDIVIOR INC. f/k/a RECKITT BENCKISER : PHARMACEUTICALS, INC., et al. : : Defendants. : __________________________________________: Goldberg, J. November 24, 2020 MEMORANDUM Defendant Reckitt Benckiser, Inc. (“Defendant”) manufactures Suboxone, a drug commonly used to combat opioid addiction.1 Suboxone previously came in tablet form, but in 2010, citing safety concerns, Defendant effectuated a change in the administration of this drug, switching from tablet to sublingual film. Various purchasers/consumers of Suboxone claimed that this switch was anticompetitive and solely designed to maintain Defendant’s market exclusivity—a scheme known as a “product hop.” These claims have resulted in multi-district, antitrust litigation before this Court. 1 Reckitt is currently known as Indivior, Inc. In December 2014, Reckitt Benckiser Pharmaceuticals, Inc. was demerged from its prior parent, the Reckitt Benckiser Group PLC, into Indivior PLC. Although Indivior is technically the named defendant in this case, the pleadings and many of the relevant exhibits use the name “Reckitt.” As discovery and class certification litigation have come to a close, the parties have raised numerous challenges under Daubert v. Merrell Dow Pharmaceuticals, 509 U.S. 579 (1993), seeking exclusion of all or selected portions of nine expert witnesses anticipated opinions. This Opinion explains my reasoning for the resolution of these motions and will hopefully set forth a clearer path towards trial. -

FREDERICK ('ERIC') RANDALL SMITH Bsc, Phd(Edin), FRSC Eric
FREDERICK ('ERIC') RANDALL SMITH BSc, PhD(Edin), FRSC Eric Smith was responsible for research activities in Duncan Flockhart Ltd, T & H Smith Ltd and Edinburgh Pharmaceutical Industries Ltd during the period 1947 to 1971. He and his colleagues made several lasting contributions to medicine and commerce notable amongst which were the creation of a new opiate industry based on large scale poppy growing in Australia, the establishment of dihydrocodeine ('DF118') as a major analgesic, and the discovery of 'Bitrex', an intensely bitter substance, now a widely used denaturant. He was elected a Fellow of the Society in 1962 and served on Council 1969-71. Born in Edinburgh on April 4th, 1911, Eric spent his childhood, his school and university days, and most of his working life and retirement in or near to the City. He died there on November 4th, 1994. Eric's enquiring attitude to life owed much to his father. This unusual man earned his living in the Actuarial Department of the Scottish Provident Institution but his main interests were astronomy and art. He was an FRAS and his personal observations led to his name being given to a minor lunar crater, and a good enough artist for his works to be hung in the Royal Academies in Edinburgh and London. Perhaps this is why Eric chose to be a scientist and his sister an art teacher. Eric was educated at the Edinburgh Institution (later named Melville College) where he was an excellent scholar and a good enough sportsman to play for the 1st XV and the FPs. -

Morphinone Reductase for the Preparation Of
Europäisches Patentamt *EP000649465B1* (19) European Patent Office Office européen des brevets (11) EP 0 649 465 B1 (12) EUROPEAN PATENT SPECIFICATION (45) Date of publication and mention (51) Int Cl.7: C12N 9/02, C12P 17/18, of the grant of the patent: C12Q 1/32 24.07.2002 Bulletin 2002/30 (86) International application number: (21) Application number: 93913236.1 PCT/GB93/01129 (22) Date of filing: 28.05.1993 (87) International publication number: WO 94/00565 (06.01.1994 Gazette 1994/02) (54) MORPHINONE REDUCTASE FOR THE PREPARATION OF HYDROMORPHONE AND HYDROCODONE MORPHINONE REDUKTASE ZUR HERSTELLUNG VON HYDROMORPHONE UND HYDROCODONE MORPHINONE REDUCTASE DESTINEE A LA PREPARATION D’HYDROMORPHONE ET D’HYDROCODONE (84) Designated Contracting States: (74) Representative: Perry, Robert Edward AT BE CH DE DK ES FR GB IE IT LI NL PT SE GILL JENNINGS & EVERY Broadgate House (30) Priority: 25.06.1992 GB 9213524 7 Eldon Street London EC2M 7LH (GB) (43) Date of publication of application: 26.04.1995 Bulletin 1995/17 (56) References cited: WO-A-90/13634 (73) Proprietor: MACFARLAN SMITH LIMITED Edinburgh EH11 2QA (GB) • THE BIOCHEMICAL JOURNAL vol. 274, no. 3, 15 March 1991, pages 875 - 880 NEIL C. BRUCE ET (72) Inventors: AL. ’Microbial degradation of the morphine • HAILES, Anne, Maria 59 Lower Road alkaloids. Purification and characterization of Kent DA8 1AY (GB) morphine dehydrogenase from Pseudomonas • FRENCH, Christopher, Edward putida M10’ Palmerston North (NZ) • BRUCE, Neil, Charles Cambridge CB2 1ND (GB) Note: Within nine months from the publication of the mention of the grant of the European patent, any person may give notice to the European Patent Office of opposition to the European patent granted. -

PHARMACEUTICALS + Biovian Oy Jemedic AB Bayer Oy + Shetland Bergen Helse Bergen HF, Haukeland 3
BARENTS SEA Jan Mayen (Norway) Tromsø NORWAY Murmansk DENMARK STRAIT NORWEGIAN SEA Raufarhöfn Bolungarvik Kalix Aromtech Oy Mosjøen SWEDEN Kemi Akureyri Luleå WHITE SEA Piteå Eskifjördur Oy Woikoski Ab + Pharmatory Ltd + Bioactive Bone Substitutes Oolu Fermion Oy Olafsvik ICELAND Skellefteå IA Hornafjördur Reykjavik 1-5 Grindavik Apotek Produktion & Laboratorier AB Oy Woikoski Ab Umeå Norrlands Universitetssjukhus Frøya Vik Trondheim Kristiansund GULF OF BOTHN Vaasa Averøya Galena Pharma Oy + Orion Oyj + Finvector Oy Sandøy Härnösand Ålesund Sundsvall Unimedic AB + MAP Medical Technologies Oy Woikoski Ab Kristinestad Tórshavn Apoteksverkiö, Framleiösluapotekiö Måløy EWOS AS Santen Oy Cytomed Oy SWEDEN - Not Shown Vitabalans Oy + Umeå + Curida AS Apotek Produktion & Laboratorier AB Oy Aga Ab Norrlands Universitetssjukhus Jemedic AB FINLAND OSLO Gävle Alby Vyborg + Nouryon Pulp & Performance Chemicals AB Hankintatukku Oy 1. Smerud Medical Research Norway AS Pcas Finland Oy Finex Oy Hamina Orion Corporation Oy Woikoski Ab 2. Catapult Life Science AS Gävle + + EUROPE - PHARMACEUTICALS + Biovian Oy Jemedic AB Bayer Oy + Shetland Bergen Helse Bergen HF, Haukeland 3. PhotoCure ASA Turku Orion Corporation A.Vogel Oy universitetssjukehus 4. GE Healthcare AS Vitabalans Oy MAP Medicals Technologies Oy + Nanoform Finland Oy Islands 5. Oncoinvent AS Fermion Oy AGA Gas AB Lumene Oy Lerwick 6. The Cyclotron Center Pharmaq AS + Scan Aqua AS Orion Corporation + 7. Oslo Universitetssykehus HF HELSINKI 8. Diatec Monoclonals AS Institute for Energy Technology -

United States Securities and Exchange Commission Form 10-K Shire Pharmaceuticals Group
UNITED STATES SECURITIES AND EXCHANGE COMMISSION WASHINGTON, D.C. 20549 FORM 10-K (Mark One) ፤ ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 For the fiscal year ended December 31, 1999 អ TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 Commission file number 0-29630 SHIRE PHARMACEUTICALS GROUP PLC (Exact name of registrant as specified in its charter) England and Wales (State or other jurisdiction (I.R.S. Employer of incorporation or organization) Identification No.) N.A. East Anton, Andover, Hampshire SP10 5RG England (Address of principal executive offices) (Zip Code) 44 1264 333455 (Registrant's telephone number, including area code) Securities registered pursuant to Section 12(b) of the Act: Title of each class Name of exchange on which registered American Depository Shares, each representing Nasdaq National Market 3 Ordinary Shares, 5 pence nominal value per share Securities registered pursuant to Section 12(g) of the Act: None (Title of class) Indicate by check mark whether the Registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the Registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ፤ No អ Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of the Registrant's knowledge, in definitive proxy or information statements incorporated by reference to Part III of this Form 10-K or any amendment to this Form 10-K. -

United States Patent Office Patented Aug
3,830,819 United States Patent Office Patented Aug. 20, 1974 2 The invention provides a process for the purification of l-dihydrocodeine containing as impurities 1-dihydrothebai 3,830,819 MANUFACTURE OF l-DHYDROCODENE none and or 1-dihydrothebainol, which comprises treating Edward Leon Grew and David Jackson Powles, Edin the impure 1-dihydrocodeine to remove said impurities burgh, Scotland, assignors to MacFarlan Smith Limited, therefrom by either extraction with an aqueous alkali or Edinburgh, Scotland 5 by passage of the material through an anion exchange No Drawing. Fied Feb. 22, 1972, Ser. No. 228,367 resin of a quaternary ammonium type in the hydroxide Claims priority, application Great Britain, Feb. 22, 1971, form whereby the proportion of said impurities is reduced. 5,081/71 The crude 1-dihydrocodeine may be one prepared by Int, C. C07d 43/28 the catalytic hydrogenation of 1-dihydrocodeinone and ac U.S. C. 260-285 3 Claims 0. cording to a feature of the invention, there is therefore provided a process for the preparation of a purified 1-di ABSTRACT OF THE DESCLOSURE hydrocodeine which comprises the steps of A process for the preparation of a purified 1-dihydro (a) catalytically hydrogenating 1-dihydrocodeinone in a codeine which comprises the steps of: 5 liquid medium using a platinum oxide or supported (a) catalytically hydrogenating 1-dihydrocodeinone in a platinum metal catalyst to produce a solution of crude liquid medium using a catalyst selected from the group l-dihydrocodeine, consisting of platinum oxide and supported platinum (b) removing the catalyst from the solution, and metal to produce a solution of crude 1-dihydrocodeine, (c) treating the solution for the recovery of a purified (b) removing the catalyst from the solution, and 20 1-dihydrocodeine having a reduced content of l-dihy (c) treating the solution for recovery of a purified 1-di drothebainone and/or 1-dihydrothebainol. -
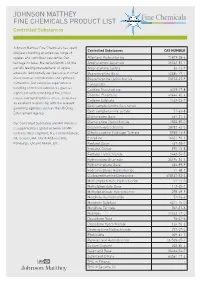
JOHNSON MATTHEY FINE CHEMICALS PRODUCT LIST Controlled Substances
JOHNSON MATTHEY FINE CHEMICALS PRODUCT LIST Controlled Substances Johnson Matthey Fine Chemicals has spent Controlled Substances CAS NUMBER 200 years building an extensive range of opiates and controlled substance. Our Alfentanil Hydrochloride 70879-28-6 heritage includes Macfarlan Smith Ltd, the Amphetamine Aspartate 25333-81-7 world’s leading manufacturer of opiate Amphetamine Sulfate 60-13-9 alkaloids. Additionally we specialise in other Buprenorphine Base 52485-79-7 areas such as cannabinoids and synthetic Buprenorphine Hydrochloride 53152-21-9 stimulants. Our extensive experience in Cannabidiol - handling controlled substances gives us Codeine Monohydrate 6059-47-8 significant understanding of the critical Codeine Phosphate 41444-62-6 issues surrounding these areas, as well as Codeine Sulphate 1420-53-7 an excellent relationship with the relevant Dextroamphetamine Saccharate - governing agencies such as the US Drug Dextroamphetamine Sulfate 51-63-8 Enforcement Agency. Diamorphine Base 561-27-3 Our Controlled Substance and API Portfolio Diamorphine Hydrochloride 1502-95-0 is supported by a global network of GMP Difenoxin Hydrochloride 28782-42-5 facilities: West Deptford, NJ; Conshohocken, Dihydrocodeine Hydrogen Tartrate 5965-13-9 PA; Devens, MA; North Andover, MA; Etorphine 14521-96-1 Edinburgh, UK and Annan, UK. Fentanyl Base 437-38-7 Fentanyl Citrate 990-73-8 Fentanyl Hydrochloride 1443-54-5 Hydrocodone Bitartrate 34195-34-1 Hydromorphone Base 466-99-9 Hydromorphone Hydrochloride 71-68-1 Lisdexamfetamine Dimesylate 608137-33-3 -

Government's Response to OFT's Review of Undertakings by Macfarlan Smith Limited
Government’s Response to OFT’s Review of Undertakings by MacFarlan Smith Limited (MSL) Market Analysis Opium in the UK – Analysis to date March 08 Sections in this report I) Background II) Home Office Undertaking III) Market Structure IV) Conclusions from Previous Market Analysis V) Taking the Analysis Forwards VI) Conclusions To Date Annex A: Copy of text from letter written by the EU Commission to EU Parliament re Opiates Annex B: Summary of responses to Home Office data request. I) Background a) OFT Review In 2006 the OFT published a report: Opium Derivatives, A Review of the Undertakings Given by Macfarlan Smith (MSL)1. This report recommended that the Government should consider competition when setting licensing policy for opium derivatives. b) Government Response The Department for Business, Enterprise and Regulatory Reform (BERR) has a general interest in OFT's market studies and co-ordinates Government responses where those studies make regulatory recommendations. Consideration of regulatory and policy issues is a matter for the relevant Department. In its response to the OFT2, the Government agreed to: Undertake to analyse the effects of MSL’s position as sole supplier on customers and competition, contrasting this with the positions of customers in other countries (both those who impose significant restrictions and those who do not). c) Work Undertaken by ERA & Drugs Liaison & Enforcement Unit (DLEU) BERR commissioned the Home Office to take forwards this market analysis. It was agreed that, at this stage, rather than repeat the OFT’s work a few years on (which was largely based on a survey of MSL’s UK customers) it made more sense to try and take the analysis forwards. -

Manufacture of Narcotic Drugs, Psychotropic Substances and Their Precursors 2013
Manufacture of Narcotic Drugs, Psychotropic Substances and their Precursors Fabrication de stupéfiants, de substances psychotropes et de leurs précurseurs Fabricación de estupefacientes, de sustancias sicotrópicas y de sus precursores Manufacture of Narcotic Drugs, Psychotropic Substances and their Precursors List of national manufacturers authorized to manufacture or convert specific narcotic drugs and psychotropic substances and of the substances actually manufactured or converted by them during 2012, and manufacturers of substances in the revised Table I of the United Nations Convention against Illicit Traffic in Narcotic Drugs and Psychotropic Substances, 1988 Fabrication de stupéfiants, de substances psychotropes et de leurs précurseurs Liste, par pays, des entreprises autorisées à fabriquer ou à transformer des stupéfiants et des substances psychotropes déterminés, et répertoire des substances effectivement fabriquées ou transformées par ces entreprises en 2012, et des fabricants de substances du Tableau I révisé de la Convention des Nations Unies contre le trafic illicite de stupéfiants et de substances psychotropes de 1988 Fabricación de estupefacientes, de sustancias sicotrópicas y de sus precursores Lista de fabricantes nacionales autorizados a fabricar o transformar determinados estupefacientes y sustancias sicotrópicas, y de sustancias efectivamente fabricadas o transformadas durante 2012, y de fabricantes de sustancias del Cuadro I revisado de la Convención de las Naciones Unidas contra el Tráfico Illícito de Estupefacientes -

Pharma &Biotech
PHARMA & BIOTECH 1/ 2016 I N Markets & Companies Innovation Manufacturing & Supply Chain Top 10 Brands in the Pharmaceuti- How the Pharma Industry Can Developing New Synthetic Routes, cal Industry, Pharma M&A Update, Bridge the Innovation Gap, Trends High-Potency Manufacturing, Drug Market Reports, Expert Opinions, and Success Factors in Chemical Shortage Prevention, Specialized Company News and Pharmaceutical Research Pharma Logistics Azelis Pharma 2016 advert 240x330_PRINT:Layout 1 02/09/2016 11:38 Page 1 Azelis Pharma – We’re here to add value to your business where it counts In a dynamic pharmaceutical market driven by constant development, changing regulations and evolving healthcare demands, you need a fully compliant partner with proven technical expertise, market knowledge and the capability to keep you more than one step ahead. Azelis serves the human, veterinary health and medical sectors across EMEA, Americas and Asia Pacifi c and represents many of the industry’s top API and excipient manufacturers. Our unrivalled product portfolio is supported by a full package of certifi cations and licenses. APIs – broad therapeutic range includes: cardiovascular, CNS, dermatology, diabetes, infections, nutrition, ophthalmology & pain Excipients – covering all functions: bases, binders, coatings, colours, diluents, fi llers, glidants, lubricants, sorbents, preservatives & sweeteners We work closely with customers to understand their formulation needs, identify new opportunities from patent and therapeutic targeting and proactively add value from product development through to manufacture. Contact us to fi nd out how Azelis can help with quality ingredient sourcing and supply. www.azelis.com · [email protected] Creating value, growing together E DITORIAL Diagnosis: Innovation Insufficiency – Treatment: Combination Therapy Since the beginning of the 21st century, pharma M&A activity has exploded. -

Oxycodone Import Policy Consultation Paper
OXYCODONE IMPORT POLICY A consultation paper OXYCODONE IMPORT POLICY A consultation paper Contents Consultation Summary 5 1. Introduction 7 2. Background 8 3. Issues 12 4. Options 14 5. The Government’s preferred option 16 6. Consultation responses 17 Annex A: Consultation StageImpact Assessment 20 3 OXYCODONE IMPORT POLICY A consultation paper Consultation Summary Scope of the consultation Topic of this This consultation focuses on the Government’s policy on the import of consultation: oxycodone to the UK, with particular reference to import for re-export. Scope of this The Government is reviewing its existing policy on the import of oxycodone to consultation: the UK. The purpose of this consultation is to re-examine the policy from first principles and gather information and opinions from stakeholders. This consultation will focus on three key areas: the UK’s obligations under the relevant UN conventions; the NHS’s requirements for a secure supply of diamorphine; and competition issues in the UK pharmaceuticals market. Geographical This policy applies to the whole of the United Kingdom. scope: Impact A consultation stage impact assessment has been prepared and can be found assessment (IA): at annex A. Basic Information To: We are particularly keen to hear from businesses or organisations directly involved in the trade in, and/or manufacture of, oxycodone. We are also keen to hear from those involved in health care who use diamorphine, and those involved in issues around drug misuse. Duration: This consultation was published on 23 November 2009. It will close on 15 February 2010. Enquiries and By post: responses: Oxycodone Consultation Drugs Licensing and Compliance Unit 4th Floor Fry Building 2 Marsham Street London SW1P 4DF By email: [email protected] Additional ways As this is a largely technical issue, of specialist interest, this will be a purely to become written exercise. -

Employment Tribunals (Scotland)
EMPLOYMENT TRIBUNALS (SCOTLAND) Case Nos: 4104948/2018 & 4104949/2018 Held in Edinburgh on 4,5,6,7 and 18 December 2018 Employment Judge: Michelle Sutherland (sitting alone) 5 Stephen Nelson Claimant Mr K McGuire of Counsel Edward Ferry Claimant 10 Mr K McGuire of Counsel Macfarlan Smith Limited Respondents Mr S Rochester, 15 Solicitor JUDGMENT OF THE EMPLOYMENT TRIBUNAL The judgement of the Tribunal is that the Claimant was not unfairly dismissed. 20 REASONS Introduction 1. The Claimant presented a complaint of unfair dismissal and sought to be re- engaged. His claim was conjoined with that of Edward Ferry, Case No. 4104949/2018. 25 2. It was agreed by the parties that the Claimant was dismissed by reason of redundancy. The Claimant was not challenging the fairness of the procedure adopted as regards the selection pool, individual and collective consultation, or the appeal process. His challenge was restricted to the Respondent’s efforts to find the Claimant alternative employment. Specifically, the issue was 30 whether the Claimant ought to have been offered work in Block 120. E.T. Z4 (WR) 4104948/2018 & 4104949/2018 Page 2 3. The Claimant gave evidence on his own behalf and led evidence from Edward Ferry, an ex-colleague. The Respondent led evidence from David Payne (Director of Manufacturing, Respondent), David Glenville (Head of Health and 5 Safety, Respondent), Keith Laing (Plant Manager, Respondent), Katy Esplin (HR Business Partner, Respondent) and Stephen Lockhart (Head of Production, Respondent). 4. The parties lodged a joint set of documents and made closing submissions. 10 Findings in Fact 5.