Aldol Reaction
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

STUDIES in NATURAL PRODUCT CHEMISTRY THESIS PRESENTED to the UNIVERSITY 0? for the DEGREE of Phd by JAMES PYOTT JOHNSTON
STUDIES IN NATURAL PRODUCT CHEMISTRY THESIS PRESENTED TO THE UNIVERSITY 0? GLASGOW FOR THE DEGREE OF PhD BY JAMES PYOTT JOHNSTON 1969 ProQuest Number: 11011932 All rights reserved INFORMATION TO ALL USERS The quality of this reproduction is dependent upon the quality of the copy submitted. In the unlikely event that the author did not send a com plete manuscript and there are missing pages, these will be noted. Also, if material had to be removed, a note will indicate the deletion. uest ProQuest 11011932 Published by ProQuest LLC(2018). Copyright of the Dissertation is held by the Author. All rights reserved. This work is protected against unauthorized copying under Title 17, United States C ode Microform Edition © ProQuest LLC. ProQuest LLC. 789 East Eisenhower Parkway P.O. Box 1346 Ann Arbor, Ml 48106- 1346 ACKTCOV/LEDGl'IEHTS • I should like, to record my sincere gratitude to the many people who have assisted me in this work during the past three years* I am particularly grateful to my supervisor, Dr* K.H* Overton, for his expert guidance and sustained interest and to Dr* G. Ferguson, who supervised the X-ray crystallographic studies* The analyses- and spectra quoted throughout are the result of the careful work carried out by the technical staff in this department and I should like, to acknowledge their important contribution* Special thanks are due to Roberta for perseverence in typing the manuscript* Thesa investigations were, performed during the tenure of a Carnegie Scholarship and I wish to express my gratitude to this body for its financial assistance and to Professor R*A. -
Stereoselective Reactions of Enolates: Auxiliaries
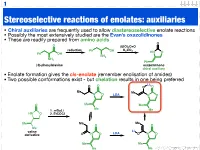
1 Stereoselective reactions of enolates: auxiliaries • Chiral auxiliaries are frequently used to allow diastereoselective enolate reactions • Possibly the most extensively studied are the Evan’s oxazolidinones • These are readily prepared from amino acids O O (EtO)2C=O reduction K CO Ph OH 2 3 HN O Ph OH NH2 NH2 Ph (S)-phenylalanine oxazolidinone chiral auxiliary • Enolate formation gives the cis-enolate (remember enolisation of amides) • Two possible conformations exist - but chelation results in one being preferred Li O O O O Me Me N O LDA N O O Me Me Me 1. n-BuLi Me HN O 2. EtCOCl Me Me Me O O Me Li valine LDA derivative O N O O N O Me Me Me Me 123.702 Organic Chemistry 2 Diastereoselective alkylation of Evan’s enolate Li O O O O Me Me N O PhCH2I N O Ph Me Me Me Me I Ph Li O Bn O O O O O H H Me N Me N H Me Me Me Me iso-propyl group blocks bottom face • Clearly (I hope) one face of the enolate is blocked • Chelation results in a rigid structure that provides maximum steric hindrance • The electrophile can only approach from one face 123.702 Organic Chemistry 3 Diastereoselective alkylation of Evan’s enolate Li O O O O Me Me N O PhCH2I N O Ph Me Me Me Me I Ph Li O Bn O O O O O H H Me N Me N H Me Me Me Me iso-propyl group blocks bottom face • Clearly (I hope) one face of the enolate is blocked • Chelation results in a rigid structure that provides maximum steric hindrance • The electrophile can only approach from one face 123.702 Organic Chemistry 4 Diastereoselective functionalisation Li O O O O Br LDA Me Me N O N O H Ph Ph 96% de H O -

Page 1 of 108 RSC Advances
RSC Advances This is an Accepted Manuscript, which has been through the Royal Society of Chemistry peer review process and has been accepted for publication. Accepted Manuscripts are published online shortly after acceptance, before technical editing, formatting and proof reading. Using this free service, authors can make their results available to the community, in citable form, before we publish the edited article. This Accepted Manuscript will be replaced by the edited, formatted and paginated article as soon as this is available. You can find more information about Accepted Manuscripts in the Information for Authors. Please note that technical editing may introduce minor changes to the text and/or graphics, which may alter content. The journal’s standard Terms & Conditions and the Ethical guidelines still apply. In no event shall the Royal Society of Chemistry be held responsible for any errors or omissions in this Accepted Manuscript or any consequences arising from the use of any information it contains. www.rsc.org/advances Page 1 of 108 RSC Advances Applications of oxazolidinones as chiral auxiliaries in the asymmetric alkylation reaction applied to total synthesis Majid M. Heravi,* Vahideh Zadsirjan, Behnaz Farajpour Department of Chemistry, School of Science, Alzahra University, Vanak, Tehran, Iran Email: [email protected] Abstract Various chiral oxazolidinones (Evans' oxazolidinones) have been employed as effective chiral auxiliaries in the asymmetric alkylation of different enolates. This strategy has been found promising and successful when used as key step (steps) in the total synthesis of several biologically active natural products. In this report, we try to underscore the applications of Manuscript oxazolidinones as chiral auxiliary in asymmetric alkylation, and particularly in crucial chiral inducing steps in the total synthesis of natural products, showing biological activities. -

Aldol Reactions: E-Enolates and Anti-Selectivity
Utah State University DigitalCommons@USU All Graduate Plan B and other Reports Graduate Studies 5-2005 Aldol Reactions: E-Enolates and Anti-Selectivity Matthew Grant Anderson Utah State University Follow this and additional works at: https://digitalcommons.usu.edu/gradreports Part of the Organic Chemistry Commons Recommended Citation Anderson, Matthew Grant, "Aldol Reactions: E-Enolates and Anti-Selectivity" (2005). All Graduate Plan B and other Reports. 1312. https://digitalcommons.usu.edu/gradreports/1312 This Report is brought to you for free and open access by the Graduate Studies at DigitalCommons@USU. It has been accepted for inclusion in All Graduate Plan B and other Reports by an authorized administrator of DigitalCommons@USU. For more information, please contact [email protected]. ALDOL REACTIONS: E-ENOLATES AND ANTI-SELECTIVITY Prepared By: MATTHEW GRANT ANDERSON A non-thesis paper submitted in partial fulfillment of the requirement for a Plan B Degree of Masters of Science in Organic Chemistry UTAH STATE UNIVERSITY Logan, Utah 2005 Contents Page CONTENTS ...................................................................................... .i LIST OF TABLES, FIGURES AND SCHEMES ....................................... ii,iii ABSTRACT .................................................................................... iv CHAPTER I. ALDOL REACTIONS:E-ENOLATES AND ANTI SELECTIVITY ......... 1 CHAPTER II. SECTION 1. MODELS OF E-ENOLATE FORMATION ...... .... ....... ... 12 SECTION 2. PATERSON ENOLATE PAPER ..... ......................... -

Robert Burns Woodward
The Life and Achievements of Robert Burns Woodward Long Literature Seminar July 13, 2009 Erika A. Crane “The structure known, but not yet accessible by synthesis, is to the chemist what the unclimbed mountain, the uncharted sea, the untilled field, the unreached planet, are to other men. The achievement of the objective in itself cannot but thrill all chemists, who even before they know the details of the journey can apprehend from their own experience the joys and elations, the disappointments and false hopes, the obstacles overcome, the frustrations subdued, which they experienced who traversed a road to the goal. The unique challenge which chemical synthesis provides for the creative imagination and the skilled hand ensures that it will endure as long as men write books, paint pictures, and fashion things which are beautiful, or practical, or both.” “Art and Science in the Synthesis of Organic Compounds: Retrospect and Prospect,” in Pointers and Pathways in Research (Bombay:CIBA of India, 1963). Robert Burns Woodward • Graduated from MIT with his Ph.D. in chemistry at the age of 20 Woodward taught by example and captivated • A tenured professor at Harvard by the age of 29 the young... “Woodward largely taught principles and values. He showed us by • Published 196 papers before his death at age example and precept that if anything is worth 62 doing, it should be done intelligently, intensely • Received 24 honorary degrees and passionately.” • Received 26 medals & awards including the -Daniel Kemp National Medal of Science in 1964, the Nobel Prize in 1965, and he was one of the first recipients of the Arthur C. -

Aldol Condensation
Chemistry 212 Laboratory Dibenzalacetone via Crossed Aldol Condensation Prelab: Calculate the amounts of all chemicals needed in measurable amounts (i.e. grams or milliliters rather than moles.) Introduction: Aldol condensations are important in organic synthesis, providing a good way to form carbon–carbon bonds. The "aldol" (aldehyde + alcohol) product is a structural unit found in many naturally occurring molecules and pharmaceuticals, and is therefore important. In an Aldol condensation an enolate ion reacts with a carbonyl compound to form a β- hydroxyaldehyde or β-hydroxyketone, followed by dehydration to give a conjugated enone. The general equation is shown in Figure 1. O O O R" B: H R R'" R "R R'" loss of H2O H R' R' Figure 1. The equation for the Aldol Condensation. The reaction involves the nucleophilic addition of an enolate to an aldehyde to form a β-hydroxy carbonyl. The β-hydroxy carbonyl is readily dehydrated under mild conditions. The aldol reaction occurs under both acidic and basic conditions as seen in Figure 2. ENOL pathway (reacts in H O protonated OH form) O O catalytic H+ O O H H R' H2O lost R' R R R' R R H aldol addition product aldol condensation product ENOLATE pathway O O M O M O base O H R' R R' R R enolate H Figure 2. The Aldol reaction and subsequent dehydration under acidic and basic conditions. The reaction we will be doing this week involves the reaction between benzaldehyde and acetone to do a double Aldol Condensation. The overall equation is shown in Figure 3. -

Collins Reagent
Collins reagent Collins reagent is the complex of chromium(VI) oxide with pyridine in dichloromethane.[2] This metal-pyridine complex, a red solid, is Collins reagent used to oxidize primary alcohols to the aldehyde. This complex is a hygroscopic orange solid.[1] Contents Names IUPAC name Synthesis and structure Pyridine - trioxochromium (2:1) Reactions Other names Other reagents Dipyridine chromium(VI) oxide[1] Safety and environmental aspects Identifiers References CAS Number 26412-88-4 (http://w ww.commonchemistr Synthesis and structure y.org/ChemicalDetai l.aspx?ref=26412-88 The complex is produced by treating chromium trioxide with -4) [2] pyridine. The complex is diamagnetic. According to X-ray 3D model Interactive image (ht (JSmol) crystallography, the complex is 5-coordinate with mutually trans tps://chemapps.stola pyridine ligands. The Cr-O and Cr-N distances are respectively 163 f.edu/jmol/jmol.php? and 215 picometers.[3] model=c1ccncc1.c1c In terms of history, the complex was first produced by Sisler et al.[4] cncc1.O%3D%5BC r%5D%28%3DO%2 Reactions 9%3DO) ChemSpider 79908 (http://www.c Collins reagent is especially useful for oxidations of acid sensitive hemspider.com/Che compounds. Primary and secondary alcohols are oxidized respectively mical-Structure.7990 to aldehydes and ketones in yields of 87-98%.[5] 8.html) Like other oxidations by Cr(VI), the stoichiometry of the oxidations is PubChem 88565 (https://pubch CID complex because the metal undergoes 3e reduction and the substrate em.ncbi.nlm.nih.gov/ is oxidized by 2 electrons: compound/88565) InChI 3 RCH2OH + 2 CrO3(pyridine)2 → 3 RCHO + 3 H2O + Cr O + 4 pyridine InChI=1S/2C5H5N.Cr.3O/c2*1-2-4-6-5-3-1;;;;/ 2 3 h2*1-5H;;;; Key: NPRDHMWYZHSAHR-UHFFFAOYSA- The reagent is typically used in a sixfold excess. -

Total Synthesis of Natural Products: a Themed Issue Dedicated to Professor Dr. Dieter Schinzer for His 65Th Birthday”
molecules Editorial Editorial to the Special Issue “Total Synthesis of Natural Products: A Themed Issue Dedicated to Professor Dr. Dieter Schinzer for His 65th Birthday” Ari M. P. Koskinen Department of Chemistry and Materials Science, Aalto University School of Chemical Engineering, Kemistintie 1, P.O. Box 16100, 02150 Espoo, Finland; ari.koskinen@aalto.fi Received: 4 December 2020; Accepted: 9 December 2020; Published: 10 December 2020 Natural products have intrigued humans throughout history. Plants with physiological activities, fermentation products, extracts with aroma, scent, or other properties have catalyzed the development of physical methods of separation of compounds and eventually the chemical synthesis of compounds. It is no coincidence that the first synthesis of an organic compound was that of a natural product. Since Wöhler’s synthesis of urea (no stereocenters) nearly two centuries ago, the synthesis of natural products has evolved through Komppa’s synthesis of camphor (one independent stereocenter) a century ago to a stage where compounds of enormous complexity can be attained through chemical synthesis (e.g., palytoxin with 64 stereocenters by Kishi in 1994). This development continues undauntedly, and it stimulates the invention of new synthetic reactions, new technological inventions, and new strategic thinking. The synthesis of natural products also stimulates the minds of medicinal chemists to develop ever better pharmaceutical products inspired by nature. Professor Dieter Schinzer had his initial training in organic synthesis with eminent mentors (Professors Manfred Reetz, Clayton Heathcock and Ekkehard Winterfeldt). Despite his wide research interests in organometallic chemistry (especially silicon, tin and manganese), synthetic methodology and medicinal chemistry, Dr. Schinzer is first and foremost a devoted natural product chemist. -

New Mesoporous Silica-Supported Organocatalysts Based on (2S)-(1,2,4-Triazol-3-Yl)-Proline: Efficient, Reusable, and Heterogeneo
molecules Article New Mesoporous Silica-Supported Organocatalysts Based on (2S)-(1,2,4-Triazol-3-yl)-Proline: Efficient, Reusable, and Heterogeneous Catalysts for the Asymmetric Aldol Reaction Omar Sánchez-Antonio 1 , Kevin A. Romero-Sedglach 1 , Erika C. Vázquez-Orta 1 and Eusebio Juaristi 1,2,* 1 Departamento de Química, Centro de Investigación y de Estudios Avanzados, Avenida IPN # 2508, 07360 Ciudad de México, Mexico; [email protected] (O.S.-A.); [email protected] (K.A.R.-S.); [email protected] (E.C.V.-O.) 2 El Colegio Nacional, Luis González Obregón # 23, Centro Histórico, 06020 Ciudad de México, Mexico * Correspondence: [email protected] or [email protected] Academic Editor: Derek J. McPhee Received: 16 September 2020; Accepted: 1 October 2020; Published: 3 October 2020 Abstract: Novel organocatalytic systems based on the recently developed (S)-proline derivative (2S)-[5-(benzylthio)-4-phenyl-(1,2,4-triazol)-3-yl]-pyrrolidine supported on mesoporous silica were prepared and their efficiency was assessed in the asymmetric aldol reaction. These materials were fully characterized by FT-IR, MS, XRD, and SEM microscopy, gathering relevant information regarding composition, morphology, and organocatalyst distribution in the doped silica. Careful optimization of the reaction conditions required for their application as catalysts in asymmetric aldol reactions between ketones and aldehydes afforded the anticipated aldol products with excellent yields and moderate diastereo- and enantioselectivities. The recommended experimental protocol is simple, fast, and efficient providing the enantioenriched aldol product, usually without the need of a special work-up or purification protocol. This approach constitutes a remarkable improvement in the field of heterogeneous (S)-proline-based organocatalysis; in particular, the solid-phase silica-bonded catalytic systems described herein allow for a substantial reduction in solvent usage. -

Tripeptide-Catalyzed Asymmetric Aldol Reaction Between Α-Ketoesters and Acetone Under Acidic Cocatalyst-Free Conditions
catalysts Article Tripeptide-Catalyzed Asymmetric Aldol Reaction Between α-ketoesters and Acetone Under Acidic Cocatalyst-Free Conditions Kazumasa Kon 1, Hiromu Takai 2, Yoshihito Kohari 3,* and Miki Murata 1,2,3 1 Graduate School of Manufacturing Engineering, Kitami Institute of Technology, 165 Koen-Cho, Kitami, Hokkaido 090-8507, Japan; [email protected] 2 Graduate School of Materials Science and Engineering, Kitami Institute of Technology, 165 Koen-Cho, Kitami, Hokkaido 090-8507, Japan; [email protected] 3 School of Earth, Energy and Environmental Engineering, Faculty of Engineering, Kitami Institute of Technology, 165 Koen-Cho, Kitami, Hokkaido 090-8507, Japan; [email protected] * Correspondence: [email protected]; Tel.: +81-157-26-9432 Received: 17 April 2019; Accepted: 6 June 2019; Published: 9 June 2019 Abstract: Here, we report the tripeptide-catalyzed asymmetric aldol reaction between α-ketoesters and acetone under acidic cocatalysts-free conditions. H-Pro-Tle-Gly-OH 3g-catalyzed reactions between α-ketoesters and acetone resulted in up to 95% yield and 88% ee. Analysis of the transition state using density functional theory (DFT) calculations revealed that the tert-butyl group in 3g played an important role in enantioselectivity. Keywords: organocatalyst; aldol reaction; peptide catalyst; α-ketoesters 1. Introduction Optically active tertiary alcohols are partial structures present in various natural products and biologically active compounds [1–5]. Various synthetic methods for these compounds have been developed. Asymmetric nucleophile addition to functionalized ketones is one of the most useful synthetic methods, because highly functionalized optically active tertiary alcohols, which can undergo various transformations, can be obtained. -

Reaction of Glucose Catalyzed by Framework and Extraframework Tin Sites in Zeolite Beta
Reaction of Glucose Catalyzed by Framework and Extraframework Tin Sites in Zeolite Beta Thesis by Ricardo Bermejo-Deval In Partial Fulfillment of the Requirements for the degree of Doctor of Philosophy CALIFORNIA INSTITUTE OF TECHNOLOGY Pasadena, California 2014 (Defended 12 May 2014) ii 2014 Ricardo Bermejo-Deval All Rights Reserved iii ACKNOWLEDGEMENTS The past years at Caltech have been one of the most challenging periods of my life, learning to be a better person and being part of the world of science. I feel privileged to have interacted with such talented and inspiring colleagues and scientists, as well as those I have disagreed with. The variety of people I have encountered and the experiences I have gathered have helped me to gain scholarship, wisdom, confidence and critical thinking, as much as to become an open-minded person to any opinion or people. First and foremost, I am very grateful to my advisor and mentor, Professor Mark E. Davis, for his financial, scientific and personal support. I appreciate his patience, flexibility, advice and scientific discussions, helping me to grow and mature scientifically. I would like to thank the thesis committee Professors Richard Flagan, Jay Labinger and Theodor Agapie, for their discussions and kindness. I appreciate everyone in the Davis group for helping me in the lab throughout these years. Special thanks to Yasho for helping me get started in the lab, to Bingjun and Raj for scientific advice, and to Marat for helpful talks. I am also very thankful to Sonjong Hwang for SS NMR, David Vandervelde for NMR and Mona Shahgholi for mass spectrometry. -

Application of Complex Aldol Reactions to the Total Synthesis of Phorboxazole B
J. Am. Chem. Soc. 2000, 122, 10033-10046 10033 Application of Complex Aldol Reactions to the Total Synthesis of Phorboxazole B David A. Evans,* Duke M. Fitch,1 Thomas E. Smith, and Victor J. Cee Contribution from the Department of Chemistry and Chemical Biology, HarVard UniVersity, Cambridge, Massachusetts 02138 ReceiVed June 29, 2000 Abstract: The synthesis of phorboxazole B has been accomplished in 27 linear steps and an overall yield of 12.6%. The absolute stereochemistry of the C4-C12,C33-C38, and C13-C19 fragments was established utilizing catalytic asymmetric aldol methodology, while the absolute stereochemistry of the C20-C32 fragment was derived from an auxiliary-based asymmetric aldol reaction. All remaining chirality was incorporated through internal asymmetric induction, with the exception of the C43 stereocenter which was derived from (R)-trityl glycidol. Key fragment couplings include a stereoselective double stereodifferentiating aldol reaction, a metalated oxazole alkylation, an oxazole-stabilized Wittig olefination, and a chelation-controlled addition of the fully elaborated alkenyl metal side chain. Introduction and HT29 (3.31 × 10-10 M), as well as leukemia CCRF-CBM (2.45 × 10-10 M), prostate cancer PC-3 (3.54 × 10-10 M), and Phorboxazoles A (1)andB(2) are marine natural products breast cancer MCF7 cell lines (5.62 × 10-10 M).2b In addition isolated from a recently discovered species of Indian Ocean to this impressive anticancer activity, phorboxazoles A and B sponge (genus Phorbas sp.) near Muiron Island, Western also exhibit potent in vitro antifungal activity against Candida Australia.2 These substances are representative of a new class albicans and Saccharomyces carlsbergensis at 0.1 µg/disk in of macrolides differing only in their C hydroxyl-bearing 13 the agar disk diffusion assay.2a stereocenters.