Atlas Journal
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Analysis of Trans Esnps Infers Regulatory Network Architecture
Analysis of trans eSNPs infers regulatory network architecture Anat Kreimer Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Graduate School of Arts and Sciences COLUMBIA UNIVERSITY 2014 © 2014 Anat Kreimer All rights reserved ABSTRACT Analysis of trans eSNPs infers regulatory network architecture Anat Kreimer eSNPs are genetic variants associated with transcript expression levels. The characteristics of such variants highlight their importance and present a unique opportunity for studying gene regulation. eSNPs affect most genes and their cell type specificity can shed light on different processes that are activated in each cell. They can identify functional variants by connecting SNPs that are implicated in disease to a molecular mechanism. Examining eSNPs that are associated with distal genes can provide insights regarding the inference of regulatory networks but also presents challenges due to the high statistical burden of multiple testing. Such association studies allow: simultaneous investigation of many gene expression phenotypes without assuming any prior knowledge and identification of unknown regulators of gene expression while uncovering directionality. This thesis will focus on such distal eSNPs to map regulatory interactions between different loci and expose the architecture of the regulatory network defined by such interactions. We develop novel computational approaches and apply them to genetics-genomics data in human. We go beyond pairwise interactions to define network motifs, including regulatory modules and bi-fan structures, showing them to be prevalent in real data and exposing distinct attributes of such arrangements. We project eSNP associations onto a protein-protein interaction network to expose topological properties of eSNPs and their targets and highlight different modes of distal regulation. -

Integrating Protein Copy Numbers with Interaction Networks to Quantify Stoichiometry in Mammalian Endocytosis
bioRxiv preprint doi: https://doi.org/10.1101/2020.10.29.361196; this version posted October 29, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. Integrating protein copy numbers with interaction networks to quantify stoichiometry in mammalian endocytosis Daisy Duan1, Meretta Hanson1, David O. Holland2, Margaret E Johnson1* 1TC Jenkins Department of Biophysics, Johns Hopkins University, 3400 N Charles St, Baltimore, MD 21218. 2NIH, Bethesda, MD, 20892. *Corresponding Author: [email protected] bioRxiv preprint doi: https://doi.org/10.1101/2020.10.29.361196; this version posted October 29, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. Abstract Proteins that drive processes like clathrin-mediated endocytosis (CME) are expressed at various copy numbers within a cell, from hundreds (e.g. auxilin) to millions (e.g. clathrin). Between cell types with identical genomes, copy numbers further vary significantly both in absolute and relative abundance. These variations contain essential information about each protein’s function, but how significant are these variations and how can they be quantified to infer useful functional behavior? Here, we address this by quantifying the stoichiometry of proteins involved in the CME network. We find robust trends across three cell types in proteins that are sub- vs super-stoichiometric in terms of protein function, network topology (e.g. -
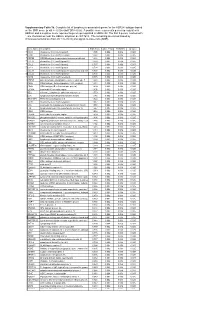
* Supplementary Table 3B. Complete List of Lymphocytic-Associated Genes
Supplementary Table 3b. Complete list of lymphocytic-associated genes for the HER2+I subtype based on the SNR score (p-val <= 0.005 and FDR<=0.05). A positive score represents genes up-regulated in HER2+I and a negative score represents genes up-regulated in HER2+NI. The first 9 genes, marked with *, are chemokines near the HER2+ amplicon at chr17q12. The remaining are sorted based by chromosomal location (from chr 1 to chr X) and signal-to-noise ratio (SNR). Gene Name Description SNR Score Feature P value FDR(BH) Q Value * CCL5 chemokine (C-C motif) ligand 5 1.395 0.002 0.036 0.025 * CCR7 chemokine (C-C motif) receptor 7 1.362 0.002 0.036 0.025 * CD79B CD79B antigen (immunoglobulin-associated beta) 1.248 0.002 0.036 0.025 * CCL13 chemokine (C-C motif) ligand 13 1.003 0.002 0.036 0.025 * CCL2 chemokine (C-C motif) ligand 2 0.737 0.004 0.056 0.037 * CCL8 chemokine (C-C motif) ligand 8 0.724 0.002 0.036 0.025 * CCL18 chemokine (C-C motif) ligand 18 (pulmonary and activ 0.703 0.002 0.036 0.025 * CCL23 chemokine (C-C motif) ligand 23 0.701 0.002 0.036 0.025 * CCR6 chemokine (C-C motif) receptor 6 0.715 0.002 0.036 0.025 PTPN7 protein tyrosine phosphatase, non-receptor type 7 1.861 0.002 0.036 0.025 CD3Z CD3Z antigen, zeta polypeptide (TiT3 complex) 1.811 0.002 0.036 0.025 CD48 CD48 antigen (B-cell membrane protein) 1.809 0.002 0.036 0.025 IL10RA interleukin 10 receptor, alpha 1.806 0.002 0.036 0.025 SELL selectin L (lymphocyte adhesion molecule 1) 1.773 0.002 0.036 0.025 LCK lymphocyte-specific protein tyrosine kinase 1.745 0.002 0.036 0.025 -

Genetic Studies of Leptin Concentrations Implicate Leptin in the Regulation of Early
Page 1 of 93 Diabetes Genetic studies of leptin concentrations implicate leptin in the regulation of early adiposity Hanieh Yaghootkar1,2,3*&, Yiying Zhang4&, Cassandra N Spracklen5, Tugce Karaderi6,7,8,9, Lam Opal Huang10, Jonathan Bradfield11,12, Claudia Schurmann13, Rebecca S Fine14,15,16, Michael H Preuss13, Zoltan Kutalik1,17,18, Laura BL Wittemans6,19, Yingchang Lu13,20, Sophia Metz10, Sara M Willems19, Ruifang Li-Gao21, Niels Grarup10, Shuai Wang22, Sophie Molnos23,24, América A Sandoval-Zárate25, Mike A Nalls26,27, Leslie A Lange28, Jeffrey Haesser29, Xiuqing Guo30, Leo-Pekka Lyytikäinen31,32, Mary F Feitosa33, Colleen M Sitlani34, Cristina Venturini35, Anubha Mahajan6,36, Tim Kacprowski37,38, Carol A Wang22, Daniel I Chasman39,40, Najaf Amin41, Linda Broer42, Neil Robertson6,36, Kristin L Young43, Matthew Allison44, Paul L Auer45, Matthias Blüher46, Judith B Borja47,48, Jette Bork-Jensen10, Germán D Carrasquilla10, Paraskevi Christofidou35, Ayse Demirkan41, Claudia A Doege49, Melissa E Garcia50, Mariaelisa Graff43,51, Kaiying Guo4, Hakon Hakonarson11,52, Jaeyoung Hong22, Yii-Der Ida Chen30, Rebecca Jackson53, Hermina Jakupović10, Pekka Jousilahti54, Anne E Justice55, Mika Kähönen56,57, Jorge R Kizer58,59, Jennifer Kriebel23,24, Charles A LeDuc4, Jin Li60, Lars Lind61, Jian’an Luan19, David Mackey62, Massimo Mangino35,63, Satu Männistö54, Jayne F Martin Carli4, Carolina Medina-Gomez41,42, Dennis O Mook-Kanamori21,64, Andrew P Morris65,6,66, Renée de Mutsert21, Matthias Nauck67,38, Ivana Nedeljkovic41, Craig E Pennell22, Arund D Pradhan39,40, Bruce M Psaty68,69, Olli T Raitakari70,71,72, Robert A Scott19, Tea Skaaby73, Konstantin Strauch74,75, Kent D Taylor30, Alexander Teumer76,38, Andre G Uitterlinden41,42, Ying Wu5, Jie Yao30, Mark Walker77, Kari E North43, Peter Kovacs46, M. -

Whole Transcriptomic Expression Differences in EBV Immortalized Versus Primary B-Cells
W&M ScholarWorks Undergraduate Honors Theses Theses, Dissertations, & Master Projects 12-2010 Whole Transcriptomic Expression Differences in EBV Immortalized versus Primary B-Cells Dolores Huberts College of William and Mary Follow this and additional works at: https://scholarworks.wm.edu/honorstheses Part of the Biology Commons Recommended Citation Huberts, Dolores, "Whole Transcriptomic Expression Differences in EBV Immortalized versus Primary B- Cells" (2010). Undergraduate Honors Theses. Paper 347. https://scholarworks.wm.edu/honorstheses/347 This Honors Thesis is brought to you for free and open access by the Theses, Dissertations, & Master Projects at W&M ScholarWorks. It has been accepted for inclusion in Undergraduate Honors Theses by an authorized administrator of W&M ScholarWorks. For more information, please contact [email protected]. Whole Transcriptomic Expression Differences in EBV Immortalized versus Primary B-Cells A thesis submitted in partial fulfillment of the requirement for the degree of Bachelor of Science with Honors in Biology from the College of William and Mary in Virginia By Dolores Huberts Accepted for Honors ________________________________________ Lizabeth A. Allison, Director ________________________________________ Matthew Wawersik ________________________________________ Drew LaMar ________________________________________ Beverly Sher Williamsburg, Virginia December 17, 2010 ABSTRACT The Epstein–Barr Virus (EBV) is a human gamma herpes virus that infects more than 90% of the human population worldwide. It is commonly known in the US as the cause of Infectious Mononucleosis, and around the world as the cause of nasopharyngeal carcinoma and malignant lymphomas such as non-Hodgkin lymphoma, endemic Burkett’s lymphoma and Hodgkin lymphoma. Additionally, the EBV is used to immortalize cells to create cell lines for in-vitro studies. -

Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name formin binding protein 1 Gene Symbol FNBP1 Organism Human Gene Summary The protein encoded by this gene is a member of the formin-binding-protein family. The protein contains an N-terminal Fer/Cdc42-interacting protein 4 (CIP4) homology (FCH) domain followed by a coiled-coil domain a proline-rich motif a second coiled-coil domain a Rho family protein-binding domain (RBD) and a C-terminal SH3 domain. This protein binds sorting nexin 2 (SNX2) tankyrase (TNKS) and dynamin; an interaction between this protein and formin has not been demonstrated yet in human. Gene Aliases FBP17, KIAA0554, MGC126804 RefSeq Accession No. NC_000009.11, NT_008470.19 UniGene ID Hs.189409 Ensembl Gene ID ENSG00000187239 Entrez Gene ID 23048 Assay Information Unique Assay ID qHsaCID0014809 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000372416, ENST00000449089, ENST00000355681, ENST00000446176, ENST00000420781, ENST00000372415 Amplicon Context Sequence GCGTGCCAAGTCCACAATGATCTGTGATGCCATGTTCTCGGAGATAACTTCATGC TGCCCTGCGTAATCATTCATTTCGTTCAGGTTGGAAATGAAAGCTTTACATGACG TATACTTGTATTCTTCTTCCTCCTTCGAGTTCTTTTTAGGTTGGTA Amplicon Length (bp) 126 Chromosome Location 9:132740777-132741637 Assay Design Intron-spanning Purification Desalted Validation Results Efficiency (%) 96 R2 0.9985 cDNA Cq 19.67 cDNA Tm (Celsius) 80.5 Page 1/5 PrimePCR™Assay Validation Report gDNA Cq 27.07 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 2/5 PrimePCR™Assay Validation -

Fibroblasts from the Human Skin Dermo-Hypodermal Junction Are
cells Article Fibroblasts from the Human Skin Dermo-Hypodermal Junction are Distinct from Dermal Papillary and Reticular Fibroblasts and from Mesenchymal Stem Cells and Exhibit a Specific Molecular Profile Related to Extracellular Matrix Organization and Modeling Valérie Haydont 1,*, Véronique Neiveyans 1, Philippe Perez 1, Élodie Busson 2, 2 1, 3,4,5,6, , Jean-Jacques Lataillade , Daniel Asselineau y and Nicolas O. Fortunel y * 1 Advanced Research, L’Oréal Research and Innovation, 93600 Aulnay-sous-Bois, France; [email protected] (V.N.); [email protected] (P.P.); [email protected] (D.A.) 2 Department of Medical and Surgical Assistance to the Armed Forces, French Forces Biomedical Research Institute (IRBA), 91223 CEDEX Brétigny sur Orge, France; [email protected] (É.B.); [email protected] (J.-J.L.) 3 Laboratoire de Génomique et Radiobiologie de la Kératinopoïèse, Institut de Biologie François Jacob, CEA/DRF/IRCM, 91000 Evry, France 4 INSERM U967, 92260 Fontenay-aux-Roses, France 5 Université Paris-Diderot, 75013 Paris 7, France 6 Université Paris-Saclay, 78140 Paris 11, France * Correspondence: [email protected] (V.H.); [email protected] (N.O.F.); Tel.: +33-1-48-68-96-00 (V.H.); +33-1-60-87-34-92 or +33-1-60-87-34-98 (N.O.F.) These authors contributed equally to the work. y Received: 15 December 2019; Accepted: 24 January 2020; Published: 5 February 2020 Abstract: Human skin dermis contains fibroblast subpopulations in which characterization is crucial due to their roles in extracellular matrix (ECM) biology. -

Retinoid X Receptor Agonists Increase Bcl2a1 Expression and Decrease Apoptosis of Naive T Lymphocytes1
The Journal of Immunology Retinoid X Receptor Agonists Increase Bcl2a1 Expression and Decrease Apoptosis of Naive T Lymphocytes1 Reuven Rasooly,* Gertrud U. Schuster,* Jeffrey P. Gregg,† Jia-Hao Xiao,‡ Roshantha A. S. Chandraratna,‡ and Charles B. Stephensen2* Vitamin A affects many aspects of T lymphocyte development and function. The vitamin A metabolites all-trans- and 9-cis-retinoic acid regulate gene expression by binding to the retinoic acid receptor (RAR), while 9-cis-retinoic acid also binds to the retinoid X receptor (RXR). Naive DO11.10 T lymphocytes expressed mRNA and protein for RAR-␣, RXR-␣, and RXR-. DNA microar- ray analysis was used to identify RXR-responsive genes in naive DO11.10 T lymphocytes treated with the RXR agonist AGN194204. A total of 128 genes was differentially expressed, including 16 (15%) involved in cell growth or apoptosis. Among these was Bcl2a1, an antiapoptotic Bcl2 family member. Quantitative real-time PCR analysis confirmed this finding and demon- strated that Bcl2a1 mRNA expression was significantly greater in nonapoptotic than in apoptotic T lymphocytes. The RXR agonist 9-cis-retinoic acid also increased Bcl2a1 expression, although all-trans-retinoic acid and ligands for other RXR partner receptors did not. Treatment with AGN194204 and 9-cis-retinoic acid significantly decreased apoptosis measured by annexin V staining but did not affect expression of Bcl2 and Bcl-xL. Bcl2a1 promoter activity was examined using a luciferase promoter construct. Both AGN194204 and 9-cis-retinoic acid significantly increased luciferase activity. In summary, these data demonstrate that RXR agonists increase Bcl2a1 promoter activity and increase expression of Bcl2a1 in naive T lymphocytes but do not affect Bcl2 and Bcl-xL expression in naive T lymphocytes. -

Anti-FBP17 / FNBP1 Antibody (ARG63662)
Product datasheet [email protected] ARG63662 Package: 100 μg anti-FBP17 / FNBP1 antibody Store at: -20°C Summary Product Description Goat Polyclonal antibody recognizes FBP17 / FNBP1 Tested Reactivity Hu Predict Reactivity Ms, Rat, Cow, Dog, Pig Tested Application IHC-P, WB Host Goat Clonality Polyclonal Isotype IgG Target Name FBP17 / FNBP1 Antigen Species Human Immunogen KQLESSKRRFERDC Conjugation Un-conjugated Alternate Names FBP17; hFBP17; Formin-binding protein 17; Formin-binding protein 1 Application Instructions Application table Application Dilution IHC-P 3 - 5 µg/ml WB 0.25 - 2 µg/ml Application Note WB: Recommend incubate at RT for 1h. IHC-P: Antigen Retrieval: Steam tissue section in Citrate buffer (pH 6.0). * The dilutions indicate recommended starting dilutions and the optimal dilutions or concentrations should be determined by the scientist. Calculated Mw 71 kDa Properties Form Liquid Purification Purified from goat serum by ammonium sulphate precipitation followed by antigen affinity chromatography using the immunizing peptide. Buffer Tris saline (pH 7.3), 0.02% Sodium azide and 0.5% BSA Preservative 0.02% Sodium azide Stabilizer 0.5% BSA Concentration 0.5 mg/ml Storage instruction For continuous use, store undiluted antibody at 2-8°C for up to a week. For long-term storage, aliquot and store at -20°C or below. Storage in frost free freezers is not recommended. Avoid repeated www.arigobio.com 1/3 freeze/thaw cycles. Suggest spin the vial prior to opening. The antibody solution should be gently mixed before use. Note For laboratory research only, not for drug, diagnostic or other use. Bioinformation Database links GeneID: 23048 Human Swiss-port # Q96RU3 Human Background The protein encoded by this gene is a member of the formin-binding-protein family. -

UC San Diego Electronic Theses and Dissertations
UC San Diego UC San Diego Electronic Theses and Dissertations Title Cardiac Stretch-Induced Transcriptomic Changes are Axis-Dependent Permalink https://escholarship.org/uc/item/7m04f0b0 Author Buchholz, Kyle Stephen Publication Date 2016 Peer reviewed|Thesis/dissertation eScholarship.org Powered by the California Digital Library University of California UNIVERSITY OF CALIFORNIA, SAN DIEGO Cardiac Stretch-Induced Transcriptomic Changes are Axis-Dependent A dissertation submitted in partial satisfaction of the requirements for the degree Doctor of Philosophy in Bioengineering by Kyle Stephen Buchholz Committee in Charge: Professor Jeffrey Omens, Chair Professor Andrew McCulloch, Co-Chair Professor Ju Chen Professor Karen Christman Professor Robert Ross Professor Alexander Zambon 2016 Copyright Kyle Stephen Buchholz, 2016 All rights reserved Signature Page The Dissertation of Kyle Stephen Buchholz is approved and it is acceptable in quality and form for publication on microfilm and electronically: Co-Chair Chair University of California, San Diego 2016 iii Dedication To my beautiful wife, Rhia. iv Table of Contents Signature Page ................................................................................................................... iii Dedication .......................................................................................................................... iv Table of Contents ................................................................................................................ v List of Figures ................................................................................................................... -

FBP17 (FNBP1) (NM 015033) Human Recombinant Protein Product Data
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for TP311011 FBP17 (FNBP1) (NM_015033) Human Recombinant Protein Product data: Product Type: Recombinant Proteins Description: Recombinant protein of human formin binding protein 1 (FNBP1) Species: Human Expression Host: HEK293T Tag: C-Myc/DDK Predicted MW: 71.1 kDa Concentration: >50 ug/mL as determined by microplate BCA method Purity: > 80% as determined by SDS-PAGE and Coomassie blue staining Buffer: 25 mM Tris.HCl, pH 7.3, 100 mM glycine, 10% glycerol Preparation: Recombinant protein was captured through anti-DDK affinity column followed by conventional chromatography steps. Storage: Store at -80°C. Stability: Stable for 12 months from the date of receipt of the product under proper storage and handling conditions. Avoid repeated freeze-thaw cycles. RefSeq: NP_055848 Locus ID: 23048 UniProt ID: Q96RU3 RefSeq Size: 5457 Cytogenetics: 9q34.11 RefSeq ORF: 1851 Synonyms: FBP17 Summary: The protein encoded by this gene is a member of the formin-binding-protein family. The protein contains an N-terminal Fer/Cdc42-interacting protein 4 (CIP4) homology (FCH) domain followed by a coiled-coil domain, a proline-rich motif, a second coiled-coil domain, a Rho family protein-binding domain (RBD), and a C-terminal SH3 domain. This protein binds sorting nexin 2 (SNX2), tankyrase (TNKS), and dynamin; an interaction between this protein and formin has not been demonstrated yet in human. [provided by RefSeq, Jul 2008] This product is to be used for laboratory only. -

Program in Human Neutrophils Fails To
Downloaded from http://www.jimmunol.org/ by guest on September 25, 2021 is online at: average * The Journal of Immunology Anaplasma phagocytophilum , 20 of which you can access for free at: 2005; 174:6364-6372; ; from submission to initial decision 4 weeks from acceptance to publication J Immunol doi: 10.4049/jimmunol.174.10.6364 http://www.jimmunol.org/content/174/10/6364 Insights into Pathogen Immune Evasion Mechanisms: Fails to Induce an Apoptosis Differentiation Program in Human Neutrophils Dori L. Borjesson, Scott D. Kobayashi, Adeline R. Whitney, Jovanka M. Voyich, Cynthia M. Argue and Frank R. DeLeo cites 28 articles Submit online. Every submission reviewed by practicing scientists ? is published twice each month by Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://jimmunol.org/subscription Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html http://www.jimmunol.org/content/suppl/2005/05/03/174.10.6364.DC1 This article http://www.jimmunol.org/content/174/10/6364.full#ref-list-1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* • Why • • Material References Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2005 by The American Association of Immunologists All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. This information is current as of September 25, 2021. The Journal of Immunology Insights into Pathogen Immune Evasion Mechanisms: Anaplasma phagocytophilum Fails to Induce an Apoptosis Differentiation Program in Human Neutrophils1 Dori L.