Genetic Studies of Leptin Concentrations Implicate Leptin in the Regulation of Early
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Analysis of Trans Esnps Infers Regulatory Network Architecture
Analysis of trans eSNPs infers regulatory network architecture Anat Kreimer Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Graduate School of Arts and Sciences COLUMBIA UNIVERSITY 2014 © 2014 Anat Kreimer All rights reserved ABSTRACT Analysis of trans eSNPs infers regulatory network architecture Anat Kreimer eSNPs are genetic variants associated with transcript expression levels. The characteristics of such variants highlight their importance and present a unique opportunity for studying gene regulation. eSNPs affect most genes and their cell type specificity can shed light on different processes that are activated in each cell. They can identify functional variants by connecting SNPs that are implicated in disease to a molecular mechanism. Examining eSNPs that are associated with distal genes can provide insights regarding the inference of regulatory networks but also presents challenges due to the high statistical burden of multiple testing. Such association studies allow: simultaneous investigation of many gene expression phenotypes without assuming any prior knowledge and identification of unknown regulators of gene expression while uncovering directionality. This thesis will focus on such distal eSNPs to map regulatory interactions between different loci and expose the architecture of the regulatory network defined by such interactions. We develop novel computational approaches and apply them to genetics-genomics data in human. We go beyond pairwise interactions to define network motifs, including regulatory modules and bi-fan structures, showing them to be prevalent in real data and exposing distinct attributes of such arrangements. We project eSNP associations onto a protein-protein interaction network to expose topological properties of eSNPs and their targets and highlight different modes of distal regulation. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Enzymatic Assay of L-METHIONINE GAMMA-LYASE (EC 4.4.1.11)
Enzymatic Assay of L-METHIONINE GAMMA-LYASE (EC 4.4.1.11) PRINCIPLE: L-Methionine Gamma-Lyase L-Methionine > Methanethiol + 2-Ketobutyrate + NH3 2-Ketobutyrate + MBTH > Azine Derivative Abbreviation used: MBTH = 3-Methyl-2-Benzothiazolinone CONDITIONS: T = 37°C, pH = 8.0, A320nm, Light path = 1 cm METHOD: Stopped Spectrophotometric Rate Determination REAGENTS: A. 100 mM Potassium Phosphate Buffer with 25 mM L-Methionine and 0.01 mM Pyridoxal 5-Phosphate, pH 8.0 at 37°C1 (Prepare 100 ml in deionized water using Potassium Phosphate, Monobasic, Anhydrous, Sigma Prod. No. P-5379, L-Methionine, Sigma Prod. No. M-9625, and Pyridoxal 5-Phosphate, Sigma Prod. No. P-9255. Adjust to pH 8.0 at 37°C with 5 M KOH.) B. 50% (w/v) Trichloroacetic Acid Solution (TCA) (Prepare 5 ml in deionized water using Trichloroacetic Acid, 6.1 N Solution, approximately 100% (w/v), Sigma Stock No. 490-10.) C. 1 M Sodium Acetate Buffer, pH 5.0 at 37°C (NaOAC) (Prepare 100 ml in deionized water using Sodium Acetate, Trihydrate, Sigma Prod. No. S-8625. Adjust to pH 5.0 at 37°C with 5 M HCl.) D. 0.1% (w/v) 3-Methyl-2-Benzothiazolinone Hydrazone (MBTH) (Prepare 10 ml in deionized water using 3-Methyl-2-Benzothiazolinone Hydrazone, Hydrochloride Hydrate, Sigma Prod. No. M-8006.) SSMETH01 Page 1 of 4 07/29/98 Enzymatic Assay of L-METHIONINE GAMMA-LYASE (EC 4.4.1.11) REAGENTS: E. 100 mM Potassium Phosphate Buffer with 1 mM Ethylenediaminetetraacetic Acid, 0.01% (v/v) 2-Mercaptoethanol and 0.02 mM Pyridoxal 5-Phosphate, pH 7.2 at 37°C (Enzyme Diluent)1 (Prepare 10 ml in deionized water using Potassium Phosphate, Monobasic, Anhydrous, Sigma Prod. -
![Lyxumia Subcutaneous Injection 300 Μg [Non-Proprietary Name] Lixisenatide (JAN*) [Name of Applicant] Sanofi K.K](https://docslib.b-cdn.net/cover/4198/lyxumia-subcutaneous-injection-300-g-non-proprietary-name-lixisenatide-jan-name-of-applicant-sanofi-k-k-324198.webp)
Lyxumia Subcutaneous Injection 300 Μg [Non-Proprietary Name] Lixisenatide (JAN*) [Name of Applicant] Sanofi K.K
Report on the Deliberation Results May 31, 2013 Evaluation and Licensing Division, Pharmaceutical and Food Safety Bureau Ministry of Health, Labour and Welfare [Brand name] Lyxumia Subcutaneous Injection 300 μg [Non-proprietary name] Lixisenatide (JAN*) [Name of applicant] Sanofi K.K. [Date of application] June 11, 2012 [Results of deliberation] In the meeting held on May 24, 2013, the First Committee on New Drugs concluded that the product may be approved and that this result should be reported to the Pharmaceutical Affairs Department of the Pharmaceutical Affairs and Food Sanitation Council. The re-examination period for the product is 8 years, and the drug substance and the drug product are both classified as powerful drugs and the product is not classified as a biological product or a specified biological product. The proposed Japanese brand name should be changed for ensuring medical safety. *Japanese Accepted Name (modified INN) This English version of the Japanese review report is intended to be a reference material to provide convenience for users. In the event of inconsistency between the Japanese original and this English translation, the former shall prevail. The PMDA will not be responsible for any consequence resulting from the use of this English version. Review Report May 7, 2013 Pharmaceuticals and Medical Devices Agency The results of a regulatory review conducted by the Pharmaceuticals and Medical Devices Agency on the following pharmaceutical product submitted for registration are as follows. [Brand name] Lyxumia Subcutaneous -

The Function and Evolution of C2H2 Zinc Finger Proteins and Transposons
The function and evolution of C2H2 zinc finger proteins and transposons by Laura Francesca Campitelli A thesis submitted in conformity with the requirements for the degree of Doctor of Philosophy Department of Molecular Genetics University of Toronto © Copyright by Laura Francesca Campitelli 2020 The function and evolution of C2H2 zinc finger proteins and transposons Laura Francesca Campitelli Doctor of Philosophy Department of Molecular Genetics University of Toronto 2020 Abstract Transcription factors (TFs) confer specificity to transcriptional regulation by binding specific DNA sequences and ultimately affecting the ability of RNA polymerase to transcribe a locus. The C2H2 zinc finger proteins (C2H2 ZFPs) are a TF class with the unique ability to diversify their DNA-binding specificities in a short evolutionary time. C2H2 ZFPs comprise the largest class of TFs in Mammalian genomes, including nearly half of all Human TFs (747/1,639). Positive selection on the DNA-binding specificities of C2H2 ZFPs is explained by an evolutionary arms race with endogenous retroelements (EREs; copy-and-paste transposable elements), where the C2H2 ZFPs containing a KRAB repressor domain (KZFPs; 344/747 Human C2H2 ZFPs) are thought to diversify to bind new EREs and repress deleterious transposition events. However, evidence of the gain and loss of KZFP binding sites on the ERE sequence is sparse due to poor resolution of ERE sequence evolution, despite the recent publication of binding preferences for 242/344 Human KZFPs. The goal of my doctoral work has been to characterize the Human C2H2 ZFPs, with specific interest in their evolutionary history, functional diversity, and coevolution with LINE EREs. -

The Un-Design and Design of Insulin: Structural Evolution
THE UN-DESIGN AND DESIGN OF INSULIN: STRUCTURAL EVOLUTION WITH APPLICATION TO THERAPEUTIC DESIGN By NISCHAY K. REGE Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy Department of Biochemistry Dissertation Advisor: Dr. Michael A. Weiss CASE WESTERN RESERVE UNIVERSITY August, 2018 CASE WESTERN RESERVE UNIVERSITY SCHOOL OF GRADUATE STUDIES We hereby approve the thesis/dissertation of Nischay K. Rege candidate for the degree of Doctor of Philosophy*. Committee Chair Paul Carey Committee Member Michael Weiss Committee Member Faramarz Ismail-Beigi Committee Member George Dubyak Date of Defense: June 26th, 2018 *We also certify that written approval has been obtained for any proprietary material contained therein. Dedication This thesis is dedicated to my mother, Dipti, whose constant love and faith have never failed, to my father, Kiran, who taught me of the virtue of curiosity, and to my wife, Shipra, whose kindness and companionship have given me enough strength for eight lifetimes. i Table of Contents Dedication ..................................................................................................................................... i Table of Contents ......................................................................................................................... ii List of Tables ............................................................................................................................... v List of Figures ........................................................................................................................... -

Integrating Protein Copy Numbers with Interaction Networks to Quantify Stoichiometry in Mammalian Endocytosis
bioRxiv preprint doi: https://doi.org/10.1101/2020.10.29.361196; this version posted October 29, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. Integrating protein copy numbers with interaction networks to quantify stoichiometry in mammalian endocytosis Daisy Duan1, Meretta Hanson1, David O. Holland2, Margaret E Johnson1* 1TC Jenkins Department of Biophysics, Johns Hopkins University, 3400 N Charles St, Baltimore, MD 21218. 2NIH, Bethesda, MD, 20892. *Corresponding Author: [email protected] bioRxiv preprint doi: https://doi.org/10.1101/2020.10.29.361196; this version posted October 29, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. Abstract Proteins that drive processes like clathrin-mediated endocytosis (CME) are expressed at various copy numbers within a cell, from hundreds (e.g. auxilin) to millions (e.g. clathrin). Between cell types with identical genomes, copy numbers further vary significantly both in absolute and relative abundance. These variations contain essential information about each protein’s function, but how significant are these variations and how can they be quantified to infer useful functional behavior? Here, we address this by quantifying the stoichiometry of proteins involved in the CME network. We find robust trends across three cell types in proteins that are sub- vs super-stoichiometric in terms of protein function, network topology (e.g. -

Atlas Journal
Atlas of Genetics and Cytogenetics in Oncology and Haematology Home Genes Leukemias Solid Tumours Cancer-Prone Deep Insight Portal Teaching X Y 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 NA Atlas Journal Atlas Journal versus Atlas Database: the accumulation of the issues of the Journal constitutes the body of the Database/Text-Book. TABLE OF CONTENTS Volume 5, Number 2, Apr-Jun 2001 Previous Issue / Next Issue Genes COL1A1 (collagen, type I, alpha 1) (17q21.31-q22). Marie-Pierre Simon, Georges Maire, Florence Pedeutour. Atlas Genet Cytogenet Oncol Haematol 2001; 5 (2): 169-177. [Full Text] [PDF] URL : http://AtlasGeneticsOncology.org/Genes/COL1A1ID186.html NF2 (neurofibromatosis type 2) (22q12.1-12.2) - updated. James F Gusella. Atlas Genet Cytogenet Oncol Haematol 2001; 5 (2): 178-184. [Full Text] [PDF] URL : http://AtlasGeneticsOncology.org/Genes/NF2117.html PDGFB (22q12.3-q13.1). Marie-Pierre Simon, Georges Maire, Florence Pedeutour. Atlas Genet Cytogenet Oncol Haematol 2001; 5 (2): 185-194. [Full Text] [PDF] URL : http://AtlasGeneticsOncology.org/Genes/PDGFBID155.html XPA (9q22.3-9q22.3). Anne Stary, Alain Sarasin. Atlas Genet Cytogenet Oncol Haematol 2001; 5 (2): 195-204. [Full Text] [PDF] Atlas Genet Cytogenet Oncol Haematol 2001; 2 I URL : http://AtlasGeneticsOncology.org/Genes/XPAID104.html ERCC-3 (Excision repair cross-complementing rodent repair deficiency, complementation group 3) (2q21). Anne Stary, Alain Sarasin. Atlas Genet Cytogenet Oncol Haematol 2001; 5 (2): 205-214. [Full Text] [PDF] URL : http://AtlasGeneticsOncology.org/Genes/XPBID296.html XPC (3p25.1). -
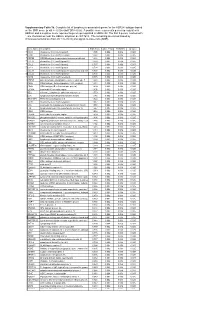
* Supplementary Table 3B. Complete List of Lymphocytic-Associated Genes
Supplementary Table 3b. Complete list of lymphocytic-associated genes for the HER2+I subtype based on the SNR score (p-val <= 0.005 and FDR<=0.05). A positive score represents genes up-regulated in HER2+I and a negative score represents genes up-regulated in HER2+NI. The first 9 genes, marked with *, are chemokines near the HER2+ amplicon at chr17q12. The remaining are sorted based by chromosomal location (from chr 1 to chr X) and signal-to-noise ratio (SNR). Gene Name Description SNR Score Feature P value FDR(BH) Q Value * CCL5 chemokine (C-C motif) ligand 5 1.395 0.002 0.036 0.025 * CCR7 chemokine (C-C motif) receptor 7 1.362 0.002 0.036 0.025 * CD79B CD79B antigen (immunoglobulin-associated beta) 1.248 0.002 0.036 0.025 * CCL13 chemokine (C-C motif) ligand 13 1.003 0.002 0.036 0.025 * CCL2 chemokine (C-C motif) ligand 2 0.737 0.004 0.056 0.037 * CCL8 chemokine (C-C motif) ligand 8 0.724 0.002 0.036 0.025 * CCL18 chemokine (C-C motif) ligand 18 (pulmonary and activ 0.703 0.002 0.036 0.025 * CCL23 chemokine (C-C motif) ligand 23 0.701 0.002 0.036 0.025 * CCR6 chemokine (C-C motif) receptor 6 0.715 0.002 0.036 0.025 PTPN7 protein tyrosine phosphatase, non-receptor type 7 1.861 0.002 0.036 0.025 CD3Z CD3Z antigen, zeta polypeptide (TiT3 complex) 1.811 0.002 0.036 0.025 CD48 CD48 antigen (B-cell membrane protein) 1.809 0.002 0.036 0.025 IL10RA interleukin 10 receptor, alpha 1.806 0.002 0.036 0.025 SELL selectin L (lymphocyte adhesion molecule 1) 1.773 0.002 0.036 0.025 LCK lymphocyte-specific protein tyrosine kinase 1.745 0.002 0.036 0.025 -

Whole Transcriptomic Expression Differences in EBV Immortalized Versus Primary B-Cells
W&M ScholarWorks Undergraduate Honors Theses Theses, Dissertations, & Master Projects 12-2010 Whole Transcriptomic Expression Differences in EBV Immortalized versus Primary B-Cells Dolores Huberts College of William and Mary Follow this and additional works at: https://scholarworks.wm.edu/honorstheses Part of the Biology Commons Recommended Citation Huberts, Dolores, "Whole Transcriptomic Expression Differences in EBV Immortalized versus Primary B- Cells" (2010). Undergraduate Honors Theses. Paper 347. https://scholarworks.wm.edu/honorstheses/347 This Honors Thesis is brought to you for free and open access by the Theses, Dissertations, & Master Projects at W&M ScholarWorks. It has been accepted for inclusion in Undergraduate Honors Theses by an authorized administrator of W&M ScholarWorks. For more information, please contact [email protected]. Whole Transcriptomic Expression Differences in EBV Immortalized versus Primary B-Cells A thesis submitted in partial fulfillment of the requirement for the degree of Bachelor of Science with Honors in Biology from the College of William and Mary in Virginia By Dolores Huberts Accepted for Honors ________________________________________ Lizabeth A. Allison, Director ________________________________________ Matthew Wawersik ________________________________________ Drew LaMar ________________________________________ Beverly Sher Williamsburg, Virginia December 17, 2010 ABSTRACT The Epstein–Barr Virus (EBV) is a human gamma herpes virus that infects more than 90% of the human population worldwide. It is commonly known in the US as the cause of Infectious Mononucleosis, and around the world as the cause of nasopharyngeal carcinoma and malignant lymphomas such as non-Hodgkin lymphoma, endemic Burkett’s lymphoma and Hodgkin lymphoma. Additionally, the EBV is used to immortalize cells to create cell lines for in-vitro studies. -
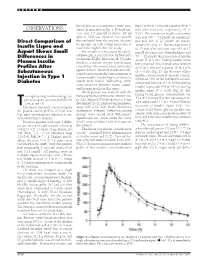
OBSERVATIONS Sured, As Indicated in Fig
LETTERS blood glucose concentrations were mea- lispro at 40 Ϯ 3 min and aspart at 49 Ϯ 3 OBSERVATIONS sured, as indicated in Fig. 1. If blood glu- min after injection, respectively (P ϭ cose was 3.5 mmol/l or lower, 20 ml 0.01). The maximum insulin concentra- glucose 30% was injected. One patient tion was 316 Ϯ 31 pmol/l on insulin lis- Direct Comparison of was excluded from the analysis because, pro and 295 Ϯ 27 pmol/l on insulin by mistake, he took a large extra dose of aspart (NS) (Fig. 1). The increase from 0 Insulin Lispro and insulin the night before the study. to 15 min after injection was 109 Ϯ 17 Aspart Shows Small Free insulin was measured after poly- pmol/l after injection of insulin lispro and Differences in ethylene glycol precipitation by Mercodia 53 Ϯ 11 pmol/l after injection of insulin Iso-Insulin (ELISA; Mercodia AB, Uppsala, Plasma Insulin aspart (P ϭ 0.02). Fasting insulin lispro Sweden), a two-site enzyme immunoassay levels reached 50% of peak concentration Profiles After containing two monoclonal antibodies at 20 Ϯ 1 min and aspart at 30 Ϯ 3 min against insulin. Identical results were ob- (P ϭ 0.02) (Fig. 2). The decrease of free Subcutaneous tained when equimolar concentrations of Injection in Type 1 insulin concentration from peak concen- human insulin, insulin lispro, and insulin tration to 50% of the maximum concen- aspart were tested, indicating 100% Diabetes tration was found at 113 Ϯ 10 min during cross-reactivity between lispro, aspart, insulin lispro and 154 Ϯ 14 min during and human insulin in this assay. -

Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name formin binding protein 1 Gene Symbol FNBP1 Organism Human Gene Summary The protein encoded by this gene is a member of the formin-binding-protein family. The protein contains an N-terminal Fer/Cdc42-interacting protein 4 (CIP4) homology (FCH) domain followed by a coiled-coil domain a proline-rich motif a second coiled-coil domain a Rho family protein-binding domain (RBD) and a C-terminal SH3 domain. This protein binds sorting nexin 2 (SNX2) tankyrase (TNKS) and dynamin; an interaction between this protein and formin has not been demonstrated yet in human. Gene Aliases FBP17, KIAA0554, MGC126804 RefSeq Accession No. NC_000009.11, NT_008470.19 UniGene ID Hs.189409 Ensembl Gene ID ENSG00000187239 Entrez Gene ID 23048 Assay Information Unique Assay ID qHsaCID0014809 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000372416, ENST00000449089, ENST00000355681, ENST00000446176, ENST00000420781, ENST00000372415 Amplicon Context Sequence GCGTGCCAAGTCCACAATGATCTGTGATGCCATGTTCTCGGAGATAACTTCATGC TGCCCTGCGTAATCATTCATTTCGTTCAGGTTGGAAATGAAAGCTTTACATGACG TATACTTGTATTCTTCTTCCTCCTTCGAGTTCTTTTTAGGTTGGTA Amplicon Length (bp) 126 Chromosome Location 9:132740777-132741637 Assay Design Intron-spanning Purification Desalted Validation Results Efficiency (%) 96 R2 0.9985 cDNA Cq 19.67 cDNA Tm (Celsius) 80.5 Page 1/5 PrimePCR™Assay Validation Report gDNA Cq 27.07 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 2/5 PrimePCR™Assay Validation