GABAB-Receptor Agonist-Based Immunotherapy for Type 1 Diabetes in NOD Mice
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

1 Evidence for Gliadin Antibodies As Causative Agents in Schizophrenia
1 Evidence for gliadin antibodies as causative agents in schizophrenia. C.J.Carter PolygenicPathways, 20 Upper Maze Hill, Saint-Leonard’s on Sea, East Sussex, TN37 0LG [email protected] Tel: 0044 (0)1424 422201 I have no fax Abstract Antibodies to gliadin, a component of gluten, have frequently been reported in schizophrenia patients, and in some cases remission has been noted following the instigation of a gluten free diet. Gliadin is a highly immunogenic protein, and B cell epitopes along its entire immunogenic length are homologous to the products of numerous proteins relevant to schizophrenia (p = 0.012 to 3e-25). These include members of the DISC1 interactome, of glutamate, dopamine and neuregulin signalling networks, and of pathways involved in plasticity, dendritic growth or myelination. Antibodies to gliadin are likely to cross react with these key proteins, as has already been observed with synapsin 1 and calreticulin. Gliadin may thus be a causative agent in schizophrenia, under certain genetic and immunological conditions, producing its effects via antibody mediated knockdown of multiple proteins relevant to the disease process. Because of such homology, an autoimmune response may be sustained by the human antigens that resemble gliadin itself, a scenario supported by many reports of immune activation both in the brain and in lymphocytes in schizophrenia. Gluten free diets and removal of such antibodies may be of therapeutic benefit in certain cases of schizophrenia. 2 Introduction A number of studies from China, Norway, and the USA have reported the presence of gliadin antibodies in schizophrenia 1-5. Gliadin is a component of gluten, intolerance to which is implicated in coeliac disease 6. -

Structural Basis for Potentiation by Alcohols and Anaesthetics in a Ligand-Gated Ion Channel
ARTICLE Received 10 Jul 2012 | Accepted 28 Feb 2013 | Published 16 Apr 2013 DOI: 10.1038/ncomms2682 Structural basis for potentiation by alcohols and anaesthetics in a ligand-gated ion channel Ludovic Sauguet1,2,3,4,*, Rebecca J. Howard5,w,*, Laurie Malherbe1,2,3,4,UiS.Lee5, Pierre-Jean Corringer3,4, R. Adron Harris5 & Marc Delarue1,2 Ethanol alters nerve signalling by interacting with proteins in the central nervous system, particularly pentameric ligand-gated ion channels. A recent series of mutagenesis experi- ments on Gloeobacter violaceus ligand-gated ion channel, a prokaryotic member of this family, identified a single-site variant that is potentiated by pharmacologically relevant concentra- tions of ethanol. Here we determine crystal structures of the ethanol-sensitized variant in the absence and presence of ethanol and related modulators, which bind in a transmembrane cavity between channel subunits and may stabilize the open form of the channel. Structural and mutagenesis studies defined overlapping mechanisms of potentiation by alcohols and anaesthetics via the inter-subunit cavity. Furthermore, homology modelling show this cavity to be conserved in human ethanol-sensitive glycine and GABA(A) receptors, and to involve residues previously shown to influence alcohol and anaesthetic action on these proteins. These results suggest a common structural basis for ethanol potentiation of an important class of targets for neurological actions of ethanol. 1 Unite´ de Dynamique Structurale des Macromole´cules, Institut Pasteur, F-75015 Paris, France. 2 UMR 3258, Centre National de la Recherche Scientifique, F-75015 Paris, France. 3 Groupe Re´cepteurs-Canaux, Institut Pasteur, F-75015 Paris, France. 4 Unite´ de Recherche Associe´e 2182, Centre National de la Recherche Scientifique, F-75015 Paris, France. -

GABA Receptors
D Reviews • BIOTREND Reviews • BIOTREND Reviews • BIOTREND Reviews • BIOTREND Reviews Review No.7 / 1-2011 GABA receptors Wolfgang Froestl , CNS & Chemistry Expert, AC Immune SA, PSE Building B - EPFL, CH-1015 Lausanne, Phone: +41 21 693 91 43, FAX: +41 21 693 91 20, E-mail: [email protected] GABA Activation of the GABA A receptor leads to an influx of chloride GABA ( -aminobutyric acid; Figure 1) is the most important and ions and to a hyperpolarization of the membrane. 16 subunits with γ most abundant inhibitory neurotransmitter in the mammalian molecular weights between 50 and 65 kD have been identified brain 1,2 , where it was first discovered in 1950 3-5 . It is a small achiral so far, 6 subunits, 3 subunits, 3 subunits, and the , , α β γ δ ε θ molecule with molecular weight of 103 g/mol and high water solu - and subunits 8,9 . π bility. At 25°C one gram of water can dissolve 1.3 grams of GABA. 2 Such a hydrophilic molecule (log P = -2.13, PSA = 63.3 Å ) cannot In the meantime all GABA A receptor binding sites have been eluci - cross the blood brain barrier. It is produced in the brain by decarb- dated in great detail. The GABA site is located at the interface oxylation of L-glutamic acid by the enzyme glutamic acid decarb- between and subunits. Benzodiazepines interact with subunit α β oxylase (GAD, EC 4.1.1.15). It is a neutral amino acid with pK = combinations ( ) ( ) , which is the most abundant combi - 1 α1 2 β2 2 γ2 4.23 and pK = 10.43. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Download Download
Supplementary Figure S1. Results of flow cytometry analysis, performed to estimate CD34 positivity, after immunomagnetic separation in two different experiments. As monoclonal antibody for labeling the sample, the fluorescein isothiocyanate (FITC)- conjugated mouse anti-human CD34 MoAb (Mylteni) was used. Briefly, cell samples were incubated in the presence of the indicated MoAbs, at the proper dilution, in PBS containing 5% FCS and 1% Fc receptor (FcR) blocking reagent (Miltenyi) for 30 min at 4 C. Cells were then washed twice, resuspended with PBS and analyzed by a Coulter Epics XL (Coulter Electronics Inc., Hialeah, FL, USA) flow cytometer. only use Non-commercial 1 Supplementary Table S1. Complete list of the datasets used in this study and their sources. GEO Total samples Geo selected GEO accession of used Platform Reference series in series samples samples GSM142565 GSM142566 GSM142567 GSM142568 GSE6146 HG-U133A 14 8 - GSM142569 GSM142571 GSM142572 GSM142574 GSM51391 GSM51392 GSE2666 HG-U133A 36 4 1 GSM51393 GSM51394 only GSM321583 GSE12803 HG-U133A 20 3 GSM321584 2 GSM321585 use Promyelocytes_1 Promyelocytes_2 Promyelocytes_3 Promyelocytes_4 HG-U133A 8 8 3 GSE64282 Promyelocytes_5 Promyelocytes_6 Promyelocytes_7 Promyelocytes_8 Non-commercial 2 Supplementary Table S2. Chromosomal regions up-regulated in CD34+ samples as identified by the LAP procedure with the two-class statistics coded in the PREDA R package and an FDR threshold of 0.5. Functional enrichment analysis has been performed using DAVID (http://david.abcc.ncifcrf.gov/) -

Ligand-Gated Ion Channels' British Journal of Pharmacology, Vol
Edinburgh Research Explorer The Concise Guide to PHARMACOLOGY 2015/16 Citation for published version: Alexander, SP, Peters, JA, Kelly, E, Marrion, N, Benson, HE, Faccenda, E, Pawson, AJ, Sharman, JL, Southan, C, Davies, JA & CGTP Collaborators 2015, 'The Concise Guide to PHARMACOLOGY 2015/16: Ligand-gated ion channels' British Journal of Pharmacology, vol. 172, no. 24, pp. 5870-5903. DOI: 10.1111/bph.13350 Digital Object Identifier (DOI): 10.1111/bph.13350 Link: Link to publication record in Edinburgh Research Explorer Document Version: Publisher's PDF, also known as Version of record Published In: British Journal of Pharmacology General rights Copyright for the publications made accessible via the Edinburgh Research Explorer is retained by the author(s) and / or other copyright owners and it is a condition of accessing these publications that users recognise and abide by the legal requirements associated with these rights. Take down policy The University of Edinburgh has made every reasonable effort to ensure that Edinburgh Research Explorer content complies with UK legislation. If you believe that the public display of this file breaches copyright please contact [email protected] providing details, and we will remove access to the work immediately and investigate your claim. Download date: 05. Apr. 2019 S.P.H. Alexander et al. The Concise Guide to PHARMACOLOGY 2015/16: Ligand-gated ion channels. British Journal of Pharmacology (2015) 172, 5870–5903 THE CONCISE GUIDE TO PHARMACOLOGY 2015/16: Ligand-gated ion channels Stephen PH Alexander1, -

Characterization of Zebrafish GABAA Receptor Subunits
www.nature.com/scientificreports OPEN Characterization of zebrafsh GABAA receptor subunits Kenichiro Sadamitsu, Leona Shigemitsu, Marina Suzuki, Daishi Ito, Makoto Kashima & Hiromi Hirata* γ-Aminobutyric acid (GABA), the major inhibitory neurotransmitter in the central nervous system, exerts its efect through the activation of GABA receptors. GABAA receptors are ligand-gated chloride channels composed of fve subunit proteins. Mammals have 19 diferent GABAA receptor subunits (α1–6, β1–3, γ1–3, δ, ε, π, θ, and ρ1–3), the physiological properties of which have been assayed by electrophysiology. However, the evolutionary conservation of the physiological characteristics of diverged GABAA receptor subunits remains unclear. Zebrafsh have 23 subunits (α1, α2a, α2b, α3–5, α6a, α6b, β1–4, γ1–3, δ, π, ζ, ρ1, ρ2a, ρ2b, ρ3a, and ρ3b), but the electrophysiological properties of these subunits have not been explored. In this study, we cloned the coding sequences for zebrafsh GABAA receptor subunits and investigated their expression patterns in larval zebrafsh by whole- mount in situ hybridization. We also performed electrophysiological recordings of GABA-evoked currents from Xenopus oocytes injected with one or multiple zebrafsh GABAA receptor subunit cRNAs and calculated the half-maximal efective concentrations (EC50s) for each. Our results revealed the spatial expressions and electrophysiological GABA sensitivities of zebrafsh GABAA receptors, suggesting that the properties of GABAA receptor subunits are conserved among vertebrates. γ-Aminobutyric acid (GABA), the major inhibitory neurotransmitter in the central nervous system of vertebrates, 1 controls the excitability of neural networks mainly through GABA A receptors . Te GABAA receptor mediates two types of inhibition, known as phasic and tonic inhibition2. -

Exome Sequencing Implicates Impaired GABA Signaling and Neuronal Ion Transport in Trigeminal Neuralgia
Washington University School of Medicine Digital Commons@Becker Open Access Publications 10-23-2020 Exome sequencing implicates impaired GABA signaling and neuronal ion transport in trigeminal neuralgia Weilai Dong Yale University Sheng Chih Jin Washington University School of Medicine in St. Louis et al Follow this and additional works at: https://digitalcommons.wustl.edu/open_access_pubs Recommended Citation Dong, Weilai; Jin, Sheng Chih; and et al, ,"Exome sequencing implicates impaired GABA signaling and neuronal ion transport in trigeminal neuralgia." iScience.,. (2020). https://digitalcommons.wustl.edu/open_access_pubs/9687 This Open Access Publication is brought to you for free and open access by Digital Commons@Becker. It has been accepted for inclusion in Open Access Publications by an authorized administrator of Digital Commons@Becker. For more information, please contact [email protected]. iScience ll OPEN ACCESS Article Exome Sequencing Implicates Impaired GABA Signaling and Neuronal Ion Transport in Trigeminal Neuralgia Weilai Dong, Sheng Chih Jin, August Allocco, ..., Yves De Koninck, Richard P. Lifton, Kristopher T. Kahle [email protected] HIGHLIGHTS Genomic analysis of trigeminal neuralgia (TN) using exome sequencing Rare mutations in GABA signaling and ion transport genes are enriched in TN cases Generation of a genetic TN mouse model engineered with a patient- specific mutation Dong et al., iScience 23, 101552 October 23, 2020 ª 2020 The Authors. https://doi.org/10.1016/ j.isci.2020.101552 iScience ll OPEN ACCESS Article Exome Sequencing Implicates Impaired GABA Signaling and Neuronal Ion Transport in Trigeminal Neuralgia Weilai Dong,1,2,27 Sheng Chih Jin,3,27 August Allocco,4,27 Xue Zeng,1,2,27 Amar H. -

Differential Co-Assembly of Α1-Gabaars Associated with Epileptic Encephalopathy
Research Report: Regular Manuscript Differential co-assembly of α1-GABAARs associated with epileptic encephalopathy https://doi.org/10.1523/JNEUROSCI.2748-19.2020 Cite as: J. Neurosci 2020; 10.1523/JNEUROSCI.2748-19.2020 Received: 18 November 2019 Revised: 5 May 2020 Accepted: 6 May 2020 This Early Release article has been peer-reviewed and accepted, but has not been through the composition and copyediting processes. The final version may differ slightly in style or formatting and will contain links to any extended data. Alerts: Sign up at www.jneurosci.org/alerts to receive customized email alerts when the fully formatted version of this article is published. Copyright © 2020 the authors 1 Differential co-assembly of α1-GABAARs 2 associated with epileptic encephalopathy 3 4 5 Saad Hannan1*, Aida H. B. Affandi1, Marielle Minere1¶, Charlotte Jones1, Pollyanna Goh2, 6 Gary Warnes2, Bernt Popp3,4, Regina Trollmann5, Dean Nizetic2,6 & Trevor G. Smart1* 7 8 1. Department of Neuroscience, Physiology and Pharmacology, University College London, 9 Gower Street, London WC1E 6BT, UK 10 2. The Blizard Institute, Barts & The London School of Medicine, Queen Mary University of 11 London, 4 Newark Street, London, E1 2AT, UK 12 3. Institute of Human Genetics, University Hospital Erlangen, Friedrich-Alexander-Universität 13 Erlangen-Nürnberg (FAU), Schwabachanlage 10, 91054 Erlangen, Germany. 14 4. Institute of Human Genetics, University of Leipzig Hospitals and Clinics, Leipzig, 15 Germany. 16 5. Department of Pediatrics, Division of Neuropediatrics, -

Research Article Microarray-Based Comparisons of Ion Channel Expression Patterns: Human Keratinocytes to Reprogrammed Hipscs To
Hindawi Publishing Corporation Stem Cells International Volume 2013, Article ID 784629, 25 pages http://dx.doi.org/10.1155/2013/784629 Research Article Microarray-Based Comparisons of Ion Channel Expression Patterns: Human Keratinocytes to Reprogrammed hiPSCs to Differentiated Neuronal and Cardiac Progeny Leonhard Linta,1 Marianne Stockmann,1 Qiong Lin,2 André Lechel,3 Christian Proepper,1 Tobias M. Boeckers,1 Alexander Kleger,3 and Stefan Liebau1 1 InstituteforAnatomyCellBiology,UlmUniversity,Albert-EinsteinAllee11,89081Ulm,Germany 2 Institute for Biomedical Engineering, Department of Cell Biology, RWTH Aachen, Pauwelstrasse 30, 52074 Aachen, Germany 3 Department of Internal Medicine I, Ulm University, Albert-Einstein Allee 11, 89081 Ulm, Germany Correspondence should be addressed to Alexander Kleger; [email protected] and Stefan Liebau; [email protected] Received 31 January 2013; Accepted 6 March 2013 Academic Editor: Michael Levin Copyright © 2013 Leonhard Linta et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Ion channels are involved in a large variety of cellular processes including stem cell differentiation. Numerous families of ion channels are present in the organism which can be distinguished by means of, for example, ion selectivity, gating mechanism, composition, or cell biological function. To characterize the distinct expression of this group of ion channels we have compared the mRNA expression levels of ion channel genes between human keratinocyte-derived induced pluripotent stem cells (hiPSCs) and their somatic cell source, keratinocytes from plucked human hair. This comparison revealed that 26% of the analyzed probes showed an upregulation of ion channels in hiPSCs while just 6% were downregulated. -

Assessment of Molecular Action of Direct Gating and Allosteric Modulatory Effects of Carisoprodol (Somartm) on GABA a Receptors
Graduate Theses, Dissertations, and Problem Reports 2015 Assessment of molecular action of direct gating and allosteric modulatory effects of carisoprodol (SomaRTM) on GABA A receptors Manoj Kumar Follow this and additional works at: https://researchrepository.wvu.edu/etd Recommended Citation Kumar, Manoj, "Assessment of molecular action of direct gating and allosteric modulatory effects of carisoprodol (SomaRTM) on GABA A receptors" (2015). Graduate Theses, Dissertations, and Problem Reports. 6022. https://researchrepository.wvu.edu/etd/6022 This Dissertation is protected by copyright and/or related rights. It has been brought to you by the The Research Repository @ WVU with permission from the rights-holder(s). You are free to use this Dissertation in any way that is permitted by the copyright and related rights legislation that applies to your use. For other uses you must obtain permission from the rights-holder(s) directly, unless additional rights are indicated by a Creative Commons license in the record and/ or on the work itself. This Dissertation has been accepted for inclusion in WVU Graduate Theses, Dissertations, and Problem Reports collection by an authorized administrator of The Research Repository @ WVU. For more information, please contact [email protected]. ASSESSMENT OF MOLECULAR ACTION OF DIRECT GATING AND ALLOSTERIC MODULATORY EFFECTS OF MEPROBAMATE (MILTOWN®) ON GABAA RECEPTORS Manish Kumar, MD, MS Dissertation submitted to the School of Pharmacy at West Virginia University in partial fulfillment of Requirements -
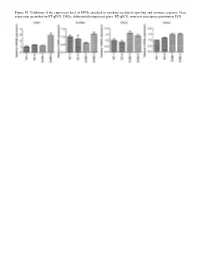
Figure S1. Validation of the Expression Level of Degs Enriched in Cytokine‑Mediated Signaling and Immune Response
Figure S1. Validation of the expression level of DEGs enriched in cytokine‑mediated signaling and immune response. Gene expression quantified by RT‑qPCR. DEGs, differentially expressed genes; RT‑qPCR, reverse‑transcription quantitative PCR. Figure S2. Validation of STAU1‑regulated AS events. IGV‑Sashimi plot revealed (A‑C) three A5SS AS events in three different genes. Reads distribution of each AS event was plotted in the left panel with the transcripts of each gene shown below. The sche‑ matic diagrams depict the structures of two AS events, AS1 (purple line) and AS2 (green line). The exon sequences are denoted by black boxes, the intron sequences by a horizontal line (right panel). RNA‑seq quantification and RT‑qPCR validation of ASEs are presented in the panels on the right. STAU1, double‑stranded RNA‑binding protein Staufen homolog 1; AS, alternative splicing; A5SS, alternative 5'splice site; RNA‑seq, RNA sequencing; RT‑qPCR, reverse‑transcription quantitative PCR. Error bars represent mean ± SEM. *P<0.05. Table SI. Primers used in gene validation experiments. IFIT2‑F CAGCCTACGGCAACTAAA IFIT2‑R GAGCCTTCTCAAAGCACA IFIT3‑F ACACCAAACAATGGCTAC IFIT3‑R TGGACAAACCCTCTAAAC OASL‑F AATGGTGACCGTGATGGG OASL‑R ACCTGAGGATGGAGCAGAG IFI27‑F TTCACTGCGGCGGGAATC IFI27‑R TGGCTGCTATGGAGGACGAG S1PR4‑F TGCTGAAGACGGTGCTGATG S1PR4‑R TGCGGAAGGAGTAGATGATGG CCL5‑F ACGACTGCTGGGTTGGAG CCL5‑R ACCCTGCTGCTTTGCCTA CCL2‑F CTAACCCAGAAACATCCAAT CCL2‑R GCTATGAGCAGCAGGCAC CD44‑F TGGAGGACAGAAAGCCAAGT CD44‑R TTCGCAATGAAACAATCAGTAG PLEKHG2‑M/As‑F CCAAAAGTAAGCCTGTCC PLEKHG2‑M‑R