(R)-Diethylene Glycol-Containing Γ-Peptide Nucleic Acids With
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Total Synthesis of Securinine and Other Methodology Studies
University of Windsor Scholarship at UWindsor Electronic Theses and Dissertations Theses, Dissertations, and Major Papers 2010 The total synthesis of securinine and other methodology studies Bhartesh Dhudshia University of Windsor Follow this and additional works at: https://scholar.uwindsor.ca/etd Recommended Citation Dhudshia, Bhartesh, "The total synthesis of securinine and other methodology studies" (2010). Electronic Theses and Dissertations. 8275. https://scholar.uwindsor.ca/etd/8275 This online database contains the full-text of PhD dissertations and Masters’ theses of University of Windsor students from 1954 forward. These documents are made available for personal study and research purposes only, in accordance with the Canadian Copyright Act and the Creative Commons license—CC BY-NC-ND (Attribution, Non-Commercial, No Derivative Works). Under this license, works must always be attributed to the copyright holder (original author), cannot be used for any commercial purposes, and may not be altered. Any other use would require the permission of the copyright holder. Students may inquire about withdrawing their dissertation and/or thesis from this database. For additional inquiries, please contact the repository administrator via email ([email protected]) or by telephone at 519-253-3000ext. 3208. The Total Synthesis of Securinine and Other Methodology Studies by Bhartesh Dhudshia A Dissertation Submitted to the Faculty of Graduate Studies through the Department of Chemistry and Biochemistry in Partial Fulfillment of the Requirements -

Benzyl Chloroformate (CHLOROFORMIC ACID, BENZYL ESTER)
Rev B Benzyl Chloroformate (CHLOROFORMIC ACID, BENZYL ESTER) C8H7O2Cl Molecular Weight = 170.6 CAS# 501‐53‐1 SPECIFICATIONS Assay: 98.% min. Color (APHA): 50 max. Benzyl Alcohol: 0.1% max. Hydrogen Chloride: 0.1% max. Benzyl Chloride: 1.5% max. Phosgene: 0.1% max. Dibenzyl Carbonate: 0.5% max. Iron: 1.5 PPM max. PHYSICAL PROPERTIES Appearance: Clear liquid free of visible contaminants BP: Decomposes at elevated temperature Odor: Pungent Density: 1.195 ‐1.22 MP/Range: ‐30°C Flash Point: 126°C NOTICE: The technical information and suggestions for use made herein are based on VanDeMark’s research and experience and are believed to be reliable, but such information and suggestions do not constitute a warranty, and no patent liability can be assumed. This publication is not to be taken as a license to operate under or infringe on any patents. Since VanDeMark has no control over the conditions under which the product is transported, stored, handled, used or applied, buyer must determine for himself by preliminary tests or otherwise, the suitability of the product for his purposes. VanDeMark’s liability on any basis is limited to the price of the product used. The information in this bulletin supersedes all previously issued bulletins on the subject matter covered. VanDeMark Benzyl Chloroformate APPLICATIONS SPILLS AND DISPOSAL Benzyl Chloroformate is a reactive chemical intermediate Use personal protective equipment (see MSDS). used in the synthesis of pharmaceutical and agrochemical Evacuate personnel to safe areas. Dike far ahead of products. It is used as a reagent in peptide synthesis to liquid spill for later disposal. -

Influence of Sulfur for Oxygen Substitution in the Solvolytic Reactions of Chloroformate Esters and Related Compounds
Int. J. Mol. Sci. 2014, 15, 18310-18332; doi:10.3390/ijms151018310 OPEN ACCESS International Journal of Molecular Sciences ISSN 1422-0067 www.mdpi.com/journal/ijms Review Influence of Sulfur for Oxygen Substitution in the Solvolytic Reactions of Chloroformate Esters and Related Compounds Malcolm J. D’Souza 1,†,* and Dennis N. Kevill 2,†,* 1 Department of Chemistry, Wesley College, 120 N. State Street, Dover, DE 19901-3875, USA 2 Department of Chemistry and Biochemistry, Northern Illinois University, DeKalb, IL 60115-2862, USA † These authors contributed equally to this work. * Authors to whom correspondence should be addressed; E-Mails: [email protected] (M.J.D.); [email protected] (D.N.K.); Tel.: +1-302-736-2528 (M.J.D.); +1-815-753-6882 (D.N.K.); Fax: +1-302-736-2301 (M.J.D.); +1-815-753-4802 (D.N.K.). External Editor: Habil. Mihai V. Putz Received: 10 September 2014; in revised form: 22 September 2014 / Accepted: 29 September 2014 / Published: 10 October 2014 Abstract: The replacement of oxygen within a chloroformate ester (ROCOCl) by sulfur can lead to a chlorothioformate (RSCOCl), a chlorothionoformate (ROCSCl), or a chlorodithioformate (RSCSCl). Phenyl chloroformate (PhOCOCl) reacts over the full range of solvents usually included in Grunwald-Winstein equation studies of solvolysis by an addition-elimination (A-E) pathway. At the other extreme, phenyl chlorodithioformate (PhSCSCl) reacts across the range by an ionization pathway. The phenyl chlorothioformate (PhSCOCl) and phenyl chlorothionoformate (PhOCSCl) react at remarkably similar rates in a given solvent and there is a dichotomy of behavior with the A-E pathway favored in solvents such as ethanol-water and the ionization mechanism favored in aqueous solvents rich in fluoroalcohol. -

Photoremovable Protecting Groups in Chemistry and Biology: Reaction Mechanisms and Efficacy Petr Klan,́*,†,‡ Tomaś̌solomek,̌ †,‡ Christian G
Review pubs.acs.org/CR Photoremovable Protecting Groups in Chemistry and Biology: Reaction Mechanisms and Efficacy Petr Klan,́*,†,‡ Tomaś̌Solomek,̌ †,‡ Christian G. Bochet,§ Aurelień Blanc,∥ Richard Givens,⊥ Marina Rubina,⊥ Vladimir Popik,# Alexey Kostikov,# and Jakob Wirz∇ † Department of Chemistry, Faculty of Science, Masaryk University, Kamenice 5, 625 00 Brno, Czech Republic ‡ Research Centre for Toxic Compounds in the Environment, Faculty of Science, Masaryk University, Kamenice 3, 625 00 Brno, Czech Republic § Department of Chemistry, University of Fribourg, Chemin du Museé 9, CH-1700 Fribourg, Switzerland ∥ Institut de Chimie, University of Strasbourg, 4 rue Blaise Pascal, 67000 Strasbourg, France ⊥ Department of Chemistry, University of Kansas, 1251 Wescoe Hall Drive, 5010 Malott Hall, Lawrence, Kansas 66045, United States # Department of Chemistry, University of Georgia, Athens, Georgia 30602, United States ∇ Department of Chemistry, University of Basel, Klingelbergstrasse 80, CH-4056 Basel, Switzerland 7.3. Arylsulfonyl Group 160 7.4. Ketones: 1,5- and 1,6-Hydrogen Abstraction 160 7.5. Carbanion-Mediated Groups 160 7.6. Sisyl and Other Silicon-Based Groups 161 7.7. 2-Hydroxycinnamyl Groups 161 7.8. α-Keto Amides, α,β-Unsaturated Anilides, CONTENTS and Methyl(phenyl)thiocarbamic Acid 162 7.9. Thiochromone S,S-Dioxide 162 1. Introduction 119 7.10. 2-Pyrrolidino-1,4-Benzoquinone Group 162 2. Arylcarbonylmethyl Groups 121 7.11. Triazine and Arylmethyleneimino Groups 162 2.1. Phenacyl and Other Related Arylcarbonyl- 7.12. Xanthene and Pyronin Groups 163 methyl Groups 121 o 7.13. Retro-Cycloaddition Reactions 163 2.2. -Alkylphenacyl Groups 123 8. Sensitized Release 163 2.3. p-Hydroxyphenacyl Groups 125 p 8.1. -
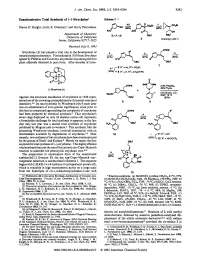
Enantioselective Total Synthesis of (-)-Strychnine1 Scheme1
J. Am. Chem. SOC.1993,115, 9293-9294 9293 Enantioselective Total Synthesis of (-)-Strychnine1 Scheme1 Steven D. Knight, Larry E. Overman,' and Garry Pairaudeau Department of Chemistry University of California Imine, California 9271 7-2025 Received July 8, 1993 OTlPS Strychnine (1) has played a vital role in the development of d h,i natural productschemistry. First isolated in 1818 fromstrychnos ignatii by Pelletier and Caventou, strychnine was among the first plant alkaloids obtained in pure form. After decades of inves- OBU' (-)-Strychnine(1) tigation, the structural elucidation of strychnine in 1946 repre- sented one of the crowning accomplishments of classical structural chemistry.394 Its total synthesis by Woodward only 8 years later was an achievement of even greater significance, since prior to this feat no compound approaching the complexity of strychnine ,oms had been prepared by chemical synthesis.* That strychnine's P' seven rings displayed on only 24 skeletal atoms still represents a formidable challenge for total synthesis is apparent in the fact that only last year was a second total synthesis of strychnine published by Magnus and co-workers.6 This synthesis, like the pioneering Woodward synthesis, involved intersection with an intermediate available by degradation of stry~hnine.~.~Most recently, two syntheses of (*)-strychnine have been communicated by the groups of Stork' and Kuehne.* Herein we report the first asymmetric total synthesis of (-)-strychnine. This highly efficient total synthesis features the use of the cationic aza-Cope-Mannich reaction to assemble the pentacyclic strychnan c~re.~J~ The preparation in enantiopure form of the unsaturated azabicyclo[3.2. -

Approach and Synthesis of Strychnos Alkaloids
N° d'ordre : 4155 THÈSE Présentée à L'UNIVERSITÉ BORDEAUX I ÉCOLE DOCTORALE DES SCIENCES CHIMIQUES par Dawood Hosni DAWOOD POUR OBTENIR LE GRADE DE DOCTEUR SPÉCIALITÉ : CHIMIE ORGANIQUE ********************* TOWARDS THE SYNTHESIS OF MONOTERPENOIDS INDOLE ALKALOIDS OF THE ASPIDOSPERMATAN AND STRYCHNAN TYPE ********************* Soutenue le: 17 décembre 2010 Après avis de: MM. PIVA Olivier Professeur, Claude Bernard Lyon 1 Rapporteur PALE Patrick Professeur, Louis Pasteur Strasbourg 1 Rapporteur Devant la commission d'examen formée de : MM. PIVA Olivier Professeur, Claude Bernard Lyon 1 Rapporteur PALE Patrick Professeur, Louis Pasteur Strasbourg 1 Rapporteur POISSON Jean-François Chargé de recherche, CNRS Examinateur VINCENT Jean-Marc Directeur de recherche, CNRS Examinateur LANDAIS Yannick Professeur, Bordeaux 1 Directeur de thèse ROBERT Frédéric Chargé de recherche, CNRS Codirecteur de thèse - 2010 - Abbreviations ∆: reflux °C: celsius degrees Ac: acetyle ALB Aluminium Lithium bis(binaphthoxide) complex AIBN : azobis(isobutyronitrile) aq.: aqueous Ar : aromatic BINAP : 2,2'-bis(diphenylphosphino)-1,1'-binaphthyle BINAPO : 2-diphenylphosphino-2'-diphenylphosphinyl-1,1'-binaphthalene BINOL: 1,1’-bi-2-naphthol Boc: tert-butyloxycarbonyle BOX: Bisoxazoline Bz : benzoyle Bn: benzyle cat. : catalytic DBU: 1,8-diazabicyclo[5.4.0]undec-7-ene DCM: dichloromethane DCC: dicyclohexacarbodiimide dr.: diastereomeric ratio DIBAL-H: diisobutylaluminium hydride DIPEA: diisopropyléthylamine (Hünig Base) DMAP: dimethylaminopyridine DME: dimethoxyethane -

United States Patent (19) 11 Patent Number: 4,760,196 Fukumoto Et Al
United States Patent (19) 11 Patent Number: 4,760,196 Fukumoto et al. 45 Date of Patent: Jul. 26, 1988 54 METHOD FOR PREPARING AN ALDEHYDE OR ALCOHOL BY REDUCTION OF A OTHER PUBLICATIONS CARBOXYLIC ACD Nystron, “J. Amer. Chem. Soc.'', vol. 69, pp. 2548 (1947). 75 Inventors: Takehiko Fukumoto; Akira Brown et al., "J. Amer. Chem. Soc.", vol. 75, p. 6263 Yamamoto, both of Niigata, Japan (1953). 73 Assignee: Shin-Etsu Chemical Co., Ltd., Tokyo, Pettit et al., “J. Organic Chem.”, vol. 27, p. 2127 (1962). Japan Primary Examiner-Werren B. Lone Attorney, Agent, or Firm-Jules E. Goldberg 21 Appl. No.: 47,612 57 ABSTRACT (22 Filed: May 8, 1987 The inventive method is particularly advantageous for the preparation of an unsaturated alcohol or aldehyde 30 Foreign Application Priority Data by the reduction of a corresponding unsaturated car boxylic acid since the unsaturated double bond in the May 12, 1986 JP Japan ................................ 61-108274 starting acid is not hydrogenated by the reaction of 51) Int. Cl* .............................................. C07C 45/54 reduction. The method comprises first reacting the 52 U.S. C. .................................... 568/484; 568/490; starting acid with an alkyl chloroformate to form an 568/864 intermediate compound by the dehydrochlorination and the intermediate compound is then reduced by 58. Field of Search ............... 568/484, 490, 485, 864, using sodium borohydride as the reducing agent. The 568/886 proportion of the aldehyde to alcohol in the product (56) References Cited mixture can be controlled by modifying the amount of U.S. PATENT DOCUMENTS the reducing agent and/or the reaction time. -
Protecting Agents
Please inquire for pricing and availability of listed products to our local sales representatives. 1 Protecting Agents Protecting groups are of vital importance in organic synthesis. In ● Acylation Reagents many cases, reaction conditions will effect multiple functionalities, Acyl protecting groups are usually used for the protection of which necessitate the blocking of several functional groups to hydroxy groups and amino groups. Acetyl (Ac), benzoyl (Bz), and afford the correct synthetic transformation. However, protecting pivaloyl (Piv) groups are commonly chosen. Pivaloyl groups is group attachment and removal requires their own conditions as often selected when non-sterically hindered hydroxyl groups well as individual chemical properties, and appropriate selection need to be selectively protected due to the Piv groups large size. of the correct protecting agents is vitally important for synthetic Generally, acyl protecting groups are stable under acidic and strategy. The most useful protecting agents generally need oxidative conditions. Acyl protecting groups are usually several key properties: deprotected under basic or reductive conditions (DIBAL, LAH, - The protecting agents must selectively react with the desired etc.). functional group requiring protection. - The protecting groups must be introduced in high yields O O O without any side reactions. CH3 CH3 - The protected functional groups should be stable under CH3 CH3 various reaction conditions. Ac group Bz group Piv group - The protecting groups must be chemoselectively deprotected O under specific conditions without deprotection of other types X R2 O of protecting groups. R1 OH 1 2 Particularly in total synthesis and for structurally complicated OH or DIBAL R O R compounds, designing the synthetic strategies frequently requires careful selection of protecting groups. -

Recent Developments on the Carbamation of Amines
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by IR@NEIST - North East Institute of Science and Technology (CSIR) Current Organic Chemistry, 2011, 15, 1593-1624 1593 Recent Developments on the Carbamation of Amines Devdutt Chaturvedi* Natural Products Chemistry Division, North-East Institute of Science and Technology (CSIR), Jorhat-785006, Assam, India. Abstract: Organic carbamates represent an important class of compounds showing various interesting properties. They find wide utility in various areas as pharmaceuticals, agrochemicals (pesticides, herbicides, insecticides, fungicides etc.), intermediates in organic synthe- sis, protection of amino group in peptide chemistry, and as linker in combinatorial chemistry etc. Classical synthesis of carbamates in- volves use of harmful reagents such as phosgene, its derivatives and carbon monoxide. Recently, various kinds of synthetic methods have been developed for the synthesis of organic carbamates. In the present review, I would like to highlight the recent developments on the synthesis of organic carbamates using variety of reagents. Keywords: Carbamation, amines, carbamates. 1. INTRODUCTION in organic synthesis [7], for the protection of amino groups in pep- tide chemistry [8], as linkers in combinatorial chemistry [9] etc. Organic carbamates are the stable class of compounds derived Functionalization of amines as carbamates offers an attractive from the unstable carbamic acid (H N-COOH) by the substitution 2 method for the generation of derivatives, which may have interest- of amino and acid ends through the various kinds of structurally ing medicinal and biological properties [10]. Organic carbamates diverse alkyl/aryl, aryl-alkyl or substituted alkyl/aryl and aryl-alkyl have been extensively used as useful synthons for the synthesis of groups, and are identified by the presence of the linkage O-CO-NH- structurally diverse synthetic intermediates/molecules of biological [1, 2]. -
Development of an Enzyme-Linked Immunosorbent Assay for Carbaryl
J. Agric. Food Chem. 1993, 41, 423-430 423 Development of an Enzyme-Linked Immunosorbent Assay for Carbaryl Maria-Pilar Marco,tJ Shirley J. Gee,? Hong M. Cheng,o Zeng Y. Liang,ll and Bruce D. Hammock',? Departments of Entomology and Environmental Toxicology, University of California, Davis, California 95616, Department of Biological Organic Chemistry, CID (CSIC), Jordi Girona 18-26, 08034 Barcelona, Spain, Institute of Zoology, Academia Sinica, Nanking, Taipei, Taiwan, and Institute of Zoology, Academia Sinica, 7 Zhongguancun Lu, Haitien, Beijing, China Seven rabbit antisera were obtained by immunization with either N-(l-naphthylacetyl)-6-aminohexanoic acid (3)or l-(l-naphthyl)-3-(5-carboxypentyl)urea(8) conjugated to keyhole limpet hemocyanin (KLH). From these sera Ab2114 (against 8KLH) was used for the optimization of an enzyme-linked immunosorbent assay (ELISA) for the determination of the insecticide carbaryl. An I50 of 2-5 ng/mL and a detection limit of 0.2 ng/mL were obtained using N-(2-naphthoyl)8-aminohexanoicacid (5) coupled to conalbumin as a coating antigen. No interference with naphthol, the main degradation product of carbaryl, was observed. An enzyme tracer was prepared by covalently linking hapten 5 with alkaline phosphatase. When the ELISA was performed using a format involving coating the plate with antibodies, an 150 of 0.4-0.6 ng/mL and a detection limit of 0.05 ng/mL were obtained. Preliminary studies with different samples showed that this immunoassay can be used for the determination of carbaryl in water, soil, body fluids, and food samples. This paper demonstrates the flexibility of using stable derivatives of a target compound such as carbaryl to generate antibodies and a sensitive ELISA for molecules containing functionalities susceptible to chemical hydrolysis and biodegradation. -
Chemistry from Nature: from Natural Products to Biorenewables Sean Joseph Riley Iowa State University
Iowa State University Capstones, Theses and Graduate Theses and Dissertations Dissertations 2011 Chemistry from Nature: From Natural Products to Biorenewables Sean Joseph Riley Iowa State University Follow this and additional works at: https://lib.dr.iastate.edu/etd Part of the Organic Chemistry Commons Recommended Citation Riley, Sean Joseph, "Chemistry from Nature: From Natural Products to Biorenewables" (2011). Graduate Theses and Dissertations. 12548. https://lib.dr.iastate.edu/etd/12548 This Dissertation is brought to you for free and open access by the Iowa State University Capstones, Theses and Dissertations at Iowa State University Digital Repository. It has been accepted for inclusion in Graduate Theses and Dissertations by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. Chemistry from nature: from natural products to biorenewables by Sean Riley A dissertation submitted to the graduate faculty in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY Major: Organic Chemistry Program of Study Committee: George A. Kraus, Major Professor William S. Jenks Gregory J. Phillips Klaus Schmidt-Rohr Arthur Winter Iowa State University Ames, Iowa 2011 ii GENERAL INTRODUCTION 1 CHAPTER 1. Synthesis of analogues to natural product salvinorin A Introduction 2 Results and Discussion 11 Experimental 19 References 31 CHAPTER 2: Synthesis of new antimicrobial compounds Introduction 33 Results and Discussion 35 Experimental 50 References 61 CHAPTER 3: Preparation of alpha olefins from fatty acids Introduction 64 Results and Discussion 65 Experimental 89 References 96 CHAPTER 4: New products from pyrones Introduction 97 Results and Discussion 101 Experimental 124 References 135 iii GENERAL CONCLUSIONS 138 ACKNOWLEDGEMENTS 139 1 GENERAL INTRODUCTION There is no chemistry without nature. -

Research.Pdf
FORMAL TOTAL SYNTHESIS OF PSEUDOPTEROXAZOLE. PROGRESS TOWARD TOTAL SYNTHESIS OF HAMIGERAN B. _______________________________________ A Dissertation presented to the Faculty of the Graduate School at the University of Missouri-Columbia _______________________________________________________ In Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy _____________________________________________________ by ZHENGXIN CAI Dr. Michael Harmata, Dissertation Supervisor JULY 2011 © Copyright by Zhengxin Cai 2011 All Rights Reserved The undersigned, appointed by the dean of the Graduate School, have examined the dissertation entitled FORMAL SYNTHESIS OF PSEUDOPTEROXAZOLE. PROGRESS TOWARD TOTAL SYNTHESIS OF HAMIGERAN B presented by Zhengxi Cai, a candidate for the degree of doctor of philosophy, and hereby certify that, in their opinion, it is worthy of acceptance. Professor Michael Harmata Professor Susan Z. Lever Professor Timothy E. Glass Professor Paul R. Sharp Professor Peter A. Tipton Dedicated to my daughter Caroline, my wife Jing and my parents. ACKNOWLEDGEMENTS I would like to thank my supervisor, Professor Michael Harmata, for his inspiration and pedagogics. My research and dissertation could not be done without his encouragement and strong support. I also would like to thank all of my committee members: Professor Susan Lever, Professor Timothy Glass, Professor Paul Sharp and Professor Peter Tipton, for their advice and comments. Thanks Carissa and Eric for detailed proofreading of my first craft. Thanks Jing for scanning NMR, IR, etc. I would like to acknowledge Dr. Wei Wycoff, for teaching and helping me on the NMR techniques and Dr. Charles Barnes for his X-ray crystallography support. Thanks Dr. Nathan for mass spectrum assistance. Thanks all of chemistry department staff, especially, Jerry Brightwell for his assistance and great patience.