Syntheses of Strychnine, Norfluorocurarine, Dehydrodesacetylretuline, and Valparicine Enabled by Intramolecular Cycloadditions of Zincke Aldehydes David B
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Methanesulfonyl Chloride
A Publication of Reliable Methods for the Preparation of Organic Compounds Working with Hazardous Chemicals The procedures in Organic Syntheses are intended for use only by persons with proper training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at http://www.nap.edu/catalog.php?record_id=12654). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices. In some articles in Organic Syntheses, chemical-specific hazards are highlighted in red “Caution Notes” within a procedure. It is important to recognize that the absence of a caution note does not imply that no significant hazards are associated with the chemicals involved in that procedure. Prior to performing a reaction, a thorough risk assessment should be carried out that includes a review of the potential hazards associated with each chemical and experimental operation on the scale that is planned for the procedure. Guidelines for carrying out a risk assessment and for analyzing the hazards associated with chemicals can be found in Chapter 4 of Prudent Practices. The procedures described in Organic Syntheses are provided as published and are conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein. -

Sodium Borohydride
Version 6.6 SAFETY DATA SHEET Revision Date 01/21/2021 Print Date 09/25/2021 SECTION 1: Identification of the substance/mixture and of the company/undertaking 1.1 Product identifiers Product name : Sodium borohydride Product Number : 452882 Brand : Aldrich CAS-No. : 16940-66-2 1.2 Relevant identified uses of the substance or mixture and uses advised against Identified uses : Laboratory chemicals, Synthesis of substances 1.3 Details of the supplier of the safety data sheet Company : Sigma-Aldrich Inc. 3050 SPRUCE ST ST. LOUIS MO 63103 UNITED STATES Telephone : +1 314 771-5765 Fax : +1 800 325-5052 1.4 Emergency telephone Emergency Phone # : 800-424-9300 CHEMTREC (USA) +1-703- 527-3887 CHEMTREC (International) 24 Hours/day; 7 Days/week SECTION 2: Hazards identification 2.1 Classification of the substance or mixture GHS Classification in accordance with 29 CFR 1910 (OSHA HCS) Chemicals which, in contact with water, emit flammable gases (Category 1), H260 Acute toxicity, Oral (Category 3), H301 Skin corrosion (Category 1B), H314 Serious eye damage (Category 1), H318 Reproductive toxicity (Category 1B), H360 For the full text of the H-Statements mentioned in this Section, see Section 16. 2.2 GHS Label elements, including precautionary statements Pictogram Signal word Danger Aldrich - 452882 Page 1 of 9 The life science business of Merck KGaA, Darmstadt, Germany operates as MilliporeSigma in the US and Canada Hazard statement(s) H260 In contact with water releases flammable gases which may ignite spontaneously. H301 Toxic if swallowed. H314 Causes severe skin burns and eye damage. H360 May damage fertility or the unborn child. -

Part I. the Total Synthesis Of
AN ABSTRACT OF THE THESIS OF Lester Percy Joseph Burton forthe degree of Doctor of Philosophy in Chemistry presentedon March 20, 1981. Title: Part 1 - The Total Synthesis of (±)-Cinnamodialand Related Drimane Sesquiterpenes Part 2 - Photochemical Activation ofthe Carboxyl Group Via NAcy1-2-thionothiazolidines Abstract approved: Redacted for privacy DT. James D. White Part I A total synthesis of the insect antifeedant(±)-cinnamodial ( ) and of the related drimanesesquiterpenes (±)-isodrimenin (67) and (±)-fragrolide (72)are described from the diene diester 49. Hydro- boration of 49 provided the C-6oxygenation and the trans ring junction in the form of alcohol 61. To confirm the stereoselectivity of the hydroboration, 61 was convertedto both (t)-isodrimenin (67) and (±)-fragrolide (72) in 3 steps. A diisobutylaluminum hydride reduction of 61 followed by a pyridiniumchlorochromate oxidation and treatment with lead tetraacetate yielded the dihydrodiacetoxyfuran102. The base induced elimination of acetic acid preceded theepoxidation and provided 106 which contains the desired hydroxy dialdehydefunctionality of cinnamodial in a protected form. The epoxide 106 was opened with methanol to yield the acetal 112. Reduction, hydrolysis and acetylation of 112 yielded (t)- cinnamodial in 19% overall yield. Part II - Various N- acyl- 2- thionothiazolidineswere prepared and photo- lysed in the presence of ethanol to provide the corresponding ethyl esters. The photochemical activation of the carboxyl function via these derivatives appears, for practical purposes, to be restricted tocases where a-keto hydrogen abstraction and subsequent ketene formation is favored by acyl substitution. Part 1 The Total Synthesis of (±)-Cinnamodial and Related Drimane Sesquiterpenes. Part 2 Photochemical Activation of the Carboxyl Group via N-Acy1-2-thionothiazolidines. -

The Total Synthesis of Securinine and Other Methodology Studies
University of Windsor Scholarship at UWindsor Electronic Theses and Dissertations Theses, Dissertations, and Major Papers 2010 The total synthesis of securinine and other methodology studies Bhartesh Dhudshia University of Windsor Follow this and additional works at: https://scholar.uwindsor.ca/etd Recommended Citation Dhudshia, Bhartesh, "The total synthesis of securinine and other methodology studies" (2010). Electronic Theses and Dissertations. 8275. https://scholar.uwindsor.ca/etd/8275 This online database contains the full-text of PhD dissertations and Masters’ theses of University of Windsor students from 1954 forward. These documents are made available for personal study and research purposes only, in accordance with the Canadian Copyright Act and the Creative Commons license—CC BY-NC-ND (Attribution, Non-Commercial, No Derivative Works). Under this license, works must always be attributed to the copyright holder (original author), cannot be used for any commercial purposes, and may not be altered. Any other use would require the permission of the copyright holder. Students may inquire about withdrawing their dissertation and/or thesis from this database. For additional inquiries, please contact the repository administrator via email ([email protected]) or by telephone at 519-253-3000ext. 3208. The Total Synthesis of Securinine and Other Methodology Studies by Bhartesh Dhudshia A Dissertation Submitted to the Faculty of Graduate Studies through the Department of Chemistry and Biochemistry in Partial Fulfillment of the Requirements -

덴드리틱 벤질 클로라이드의 효율적인 합성 Facile Synthesis of Dendritic Benzyl Chlorides from Their Alcohols W
Polymer(Korea), Vol. 31, No. 5, pp 417-421, 2007 덴드리틱 벤질 클로라이드의 효율적인 합성 이재욱F,7ㆍ한승철Fㆍ김희주ㆍ김정환ㆍ이언엽ㆍ김병기ㆍ성새름ㆍ강화신ㆍ김지현FFㆍ허도성FFF 동아대학교 화학과, F동아대학교 의생명과학과, FF동국대학교 생명화학공학과, FFF인제대학교 의생명화학과 (2007년 5월 9일 접수, 2007년 6월 29일 채택) Facile Synthesis of Dendritic Benzyl Chlorides from Their Alcohols with Methanesulfonyl Chloride/Et3N Jae Wook Lee*,7, Seung Choul Han*, Hee Joo Kim, Jung Hwan Kim, Un Yup Lee, Byoung-Ki Kim, Sae Reum Sung, Hwa-Shin Kang, Ji Hyeon Kim**, and Do-Sung Huh*** Department of Chemistry, Dong-A University, Hadan-2-dong, Busan 604-714, Korea *Department of Medical Bioscience, Dong-A University, Hadan-2-dong, Busan 604-714, Korea **Department of Chemical & Biochemical Engineering, Dongguk University, Seoul 100-715, Korea ***Department of Biomedicinal Chemistry, Inje University, Gim Hai 621-749, Korea (Received May 9, 2007; Accepted June 29, 2007) 초록: 덴드리틱 벤질 알코올을 트리에틸아민과 메탄술포닐클로라이드와 반응시켜서, 덴드리틱 벤질 클로라이드 의 효율적인 합성이 이루어졌다. 이 반응은 히드록시기의 메실화 반응과 염소화 반응의 2단계 반응으로 이루어지 는데, 중간체의 분리없이 한 반응 용기내에서 반응이 진행되는 경제적인 방법이다. Abstract: A successful rapid synthesis of dendritic benzyl chlorides from dendritic benzyl alcohols using methanesulfonyl chloride/Et3N as activating agents was described. In this method, each dendritic benzyl chloride can be prepared in one pot: no isolation of intermediate mesylated dendrons is required. The key steps in the syntheses of dendritic benzyl chlorides were the mesylation of the hydroxymethyl group followed by the chlorination by in-situ generated triethylammonium chloride. Keywords: benzylic alcohol, chlorination. Introduction vation and coupling steps in high yield. In addition, the coupling step must be very efficient to enable complete Dendrimers represent a novel type of polymeric material reaction. -
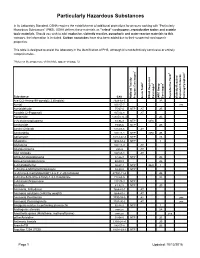
Particularly Hazardous Substances
Particularly Hazardous Substances In its Laboratory Standard, OSHA requires the establishment of additional protections for persons working with "Particularly Hazardous Substances" (PHS). OSHA defines these materials as "select" carcinogens, reproductive toxins and acutely toxic materials. Should you wish to add: explosive, violently reactive, pyrophoric and water-reactve materials to this category, the information is included. Carbon nanotubes have also been added due to their suspected carcinogenic properties. This table is designed to assist the laboratory in the identification of PHS, although it is not definitively conclusive or entirely comprehensive. *Notes on the proper use of this table appear on page 12. 1 6 5 2 3 4 Substance CAS National Toxicity National Program Carcinogen Toxin Acute Regulated OSHA Carcinogen Group IARC Carcinogen Toxin Reproductive Violently Reactive/ Explosive/Peroxide Forming/Pyrophoric A-a-C(2-Amino-9H-pyrido[2,3,b]indole) 2648-68-5 2B Acetal 105-57-7 yes Acetaldehyde 75-07-0 NTP AT 2B Acrolein (2-Propenal) 107-02-8 AT Acetamide 126850-14-4 2B 2-Acetylaminofluorene 53-96-3 NTP ORC Acrylamide 79-06-6 NTP 2B Acrylyl Chloride 814-68-6 AT Acrylonitrile 107-13-1 NTP ORC 2B Adriamycin 23214-92-8 NTP 2A Aflatoxins 1402-68-2 NTP 1 Allylamine 107-11-9 AT Alkylaluminums varies AT Allyl Chloride 107-05-1 AT ortho-Aminoazotoluene 97-56-3 NTP 2B para-aminoazobenzene 60-09-3 2B 4-Aminobiphenyl 92-67-1 NTP ORC 1 1-Amino-2-Methylanthraquinone 82-28-0 NTP (2-Amino-6-methyldipyrido[1,2-a:3’,2’-d]imidazole) 67730-11-4 2B -

The Reaction of Sodium Borohydride
THE REACTION OF SODIUM BOROHYDRIDE WITH N-·ACYLANILINES By ROBERT FRA.."\\JK LINDEl"J.ANN Ir Bachelor of Arts Wittenberg College Springfield, Ohio 1952 Submitted to the Faculty of the Graduate School of the Oklahoma Agricultural and Mechanical College in partial fulfillment of the requirements for the degree of MA.STER OF SCIENCE August, 19.54 i THE REACTION OF SODIUM BOROHYDRIDE WITH N-ACYLANILINES Thesis Approved: ~ - Dean of the Graduate School ii l!l Al C! 0~ 11J/~~. 2t"'I'Ii .ACKN01rJLEDGEMENT The author-wishes to express his gratitude to Dr. H. Po Johnston for the invaluable assistance and guidance given. This project was made possible by the Chemistry Department in the form of a Graduate Fellowship and by an unnamed sponsor through the Research Foundation. iii TABLE OF CONTENTS Page · I. .Introduction ...................................... ., o ••••••·· .... 1 II o Historical ... o o • .,, • (I .. ., •.••• G ••••.•••••••• .is •••••••• "' •. o o •••••• o '°. 2 Preparation of Sodium Borohydride ........................... 2 Physical Properties ofSodium Borohydrj.de ................... 3 Chemical Properties of · Sodium Borohydride .............. " •••• 3 III. Experimental. 0 ·• •• ·•. 0 •••• 0 ·• •••• 0. (I •••• 0 0 • 0 Q • .., ••• Q e. 0. 0. 0 D. 0 e 8 Purification of Sodium Borohydride ........................... 8 Reaction of Sodium Borohydride w:i. th Acetanilide ..........._ •• 9 Reaction of Sodium Borohydride w:i. th For.manilide oo •• oe.,. .... 27 Discussion ••• o o. o. o o o o. o ·O fl o • .e. o o ........ o. o. o o • ., o ~. o" o (Io o o o .29 IV. S11ITil11ary •• 0. Cl Ct. 0 0 0. 0. 0 -0 DO O O O C. c,.. 0. 0 4 11) 0 0 0 -0 0 0 0 -0 a O O .0 0 0 .0. -

19.8 Reduction of Aldehydes and Ketones to Alcohols
19_BRCLoudon_pgs5-0.qxd 12/9/08 11:41 AM Page 914 914 CHAPTER 19 • THE CHEMISTRY OF ALDEHYDES AND KETONES. CARBONYL-ADDITION REACTIONS 19.8 REDUCTION OF ALDEHYDES AND KETONES TO ALCOHOLS Aldehydes and ketones are reduced to alcohols with either lithium aluminum hydride, LiAlH4, or sodium borohydride, NaBH4. These reactions result in the net addition of the elements of H2 across the CAO bond. H O A H O OH 4 3 | 4 " 3 LiAlH4 ether L Li| and Al | salts (19.22) V + V + cyclobutanone lithium cyclobutanol aluminum (90% yield) hydride 4 CH3O CHA O 4 CH3OH NaBH4 LL ++ CH3OH sodium p-methoxybenzaldehyde borohydride H 4 CH3OO"C H Na|_B(OCH3)4 (19.23) LLL+ "H p-methoxybenzyl alcohol (96% yield) As these examples illustrate, reduction of an aldehyde gives a primary alcohol, and reduction of a ketone gives a secondary alcohol. Lithium aluminum hydride is one of the most useful reducing agents in organic chemistry. It serves generally as a source of H:_, the hydride ion. Because hydrogen is more electroneg- ative than aluminum (Table 1.1), the Al–H bonds of the _AlH4 ion carry a substantial fraction of the negative charge. In other words, H H reacts as if it were (19.24) H "Al_ H H "Al H _ L L L 3 "H "H The hydride ion in LiAlH4 is very basic. For this reason, LiAlH4 reacts violently with water and therefore must be used in dry solvents such as anhydrous ether and THF. 1 H 1 H Li| H "Al_ H H OH H "Al HH Li| _ OH (19.25) L L L 1 L ++hydrogenL gas 3 1 "H "H (reacts further lithium aluminum with water) hydride Like many other strong bases, the hydride ion in LiAlH4 is a good nucleophile, and LiAlH4 contains its own “built-in” Lewis acid, the lithium ion. -

Subject Index
455 Subject Index Aminohydroxylation, 364 a-Aminoketone, 281 4-Aminophenol, 18 A Aminothiophene, 158 Abnormal Claisen rearrangement, 1 a-Aminothiophenols, 184 Acrolein, 378 Ammonium ylide, 383 2-(Acylamino)-toluenes, 245 Angeli-Rimini hydroxamic acid Acylation, 100, 145, 200 synthesis, 9 Acyl azides, 98 ~-Anomer, 225 Acylium ion, 145, 149, 175, 177 Anomeric center, 211 a-Acyloxycarboxamides, 298 Anomeric effect, 135 a-Acyloxyketones, 17 ANRORC mechanism, 10 a-Acyloxythioethers, 327 Anthracenes, 51 Acyl transfer, 17, 42, 228, 298, 305, Anti-Markovnikov addition, 219 327,345 Amdt-Eistert homologation, 11 AIBN, 22, 23, 415 Aryl-acetylene, 66 Alder ene reaction, 2 Arylation, 253 Alder's endo rule, Ill 0-Aryliminoethers, 67 Aldol condensation, 3, 14, 26, 34, 2-Arylindoles, 38 69, 130, 147, 172, 305, Aryl migration, 31 340,396,412 Autoxidation, 69, 115, 118 Aldosylamine, 8 Auwers reaction, 13 Alkyl migration, 16, 132, 315, 443 Axial, 347 Alkylation, 144, 145 Azalactone, I 00 N-Aikylation, 162 Azides, 125, 330 Alkylidene carbene, 151 Azirine, 6, 7, 281 Allan-Robinson reaction, 4, 228 Azulene, 310 Allene, 119 1t-Allyl complex, 414 B Allylation, 213, 414 Baeyer-Drewson Allylstannane, 213 indigo synthesis, 14 Allylsilanes, 349 Baeyer-Villiger oxidation, 16, 53 Alper carbonylation, 6 Baker-Venkataraman Alpine-borane®, 262 rearrangement, 17 Aluminum phenolate, 149 Balz-Schiemann reaction, 354 Amadori rearrangement, 8 Bamberger rearrangement, 18 Amide acetal, 74 Bamford-Stevens reaction, 19 Amides, 28, 67,276,339, 356 Bargellini reaction, 20 Amidine, -

Fragment-Based Discovery of a New Class of Inhibitors Targeting Mycobacterial Trna Modification
Supplementary Data Fragment-based discovery of a new class of inhibitors targeting mycobacterial tRNA modification Sherine E. Thomas*1, Andrew J. Whitehouse*2, Karen Brown3,4, Juan M. Belardinelli5, Ramanuj Lahiri6, M. Daben J. Libardo7, Pooja Gupta1, Sony Malhotra8, Helena I. M. Boshoff7, Mary Jackson5, Chris Abell 2, Anthony G. Coyne2, Tom L. Blundell §1, R. Andres Floto3,4, Vitor Mendes §1 1 Department of Biochemistry, University of Cambridge, 80 Tennis Court Road, Cambridge CB2 1GA, UK. 2 Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge, CB2 1EW, UK. 3 MRC Laboratory of Molecular Biology, Francis Crick Avenue, Cambridge, CB2 0QH, U.K. 4 Cambridge Centre for Lung Infection, Royal Papworth Hospital, Cambridge, CB23 3RE, U.K. 5 Mycobacteria Research Laboratories, Department of Microbiology, Immunology and Pathology, Colorado State University, Fort Collins, Colorado, USA. 6 National Hansen’s Disease Program, Healthcare Systems Bureau, HRSA, DHHS, USA 7 Tuberculosis Research Section, Laboratory of Clinical Immunology and Microbiology, National Institute of Allergy and Infectious Disease, National Institutes of Health, 9000 Rockville Pike, Bethesda, Maryland 20892, USA. 8 Birkbeck College, University of London, Malet Street, WC1E7HX, UK * Contributed equally § To whom correspondence should be addressed Results Conservation of catalytic residues in mycobacterial TrmD A multiple sequence alignment of 5 mycobacterial TrmD enzyme sequences with 10 TrmD ortholog amino acid sequences (Figure S1) shows conservation of important catalytic residues and residues involved in tRNA recognition and methyl transfer reactions. The catalytic residues Asp169 and Arg154 are highly conserved across TrmD orthologs. Key residues such as Asp50, Gly59, His46 involved in tRNA G36 and G37 base recognition, interactions with bases at positions 38, 32 and anticodon branch of wild type tRNA respectively, are also broadly conserved. -

Benzyl Chloroformate (CHLOROFORMIC ACID, BENZYL ESTER)
Rev B Benzyl Chloroformate (CHLOROFORMIC ACID, BENZYL ESTER) C8H7O2Cl Molecular Weight = 170.6 CAS# 501‐53‐1 SPECIFICATIONS Assay: 98.% min. Color (APHA): 50 max. Benzyl Alcohol: 0.1% max. Hydrogen Chloride: 0.1% max. Benzyl Chloride: 1.5% max. Phosgene: 0.1% max. Dibenzyl Carbonate: 0.5% max. Iron: 1.5 PPM max. PHYSICAL PROPERTIES Appearance: Clear liquid free of visible contaminants BP: Decomposes at elevated temperature Odor: Pungent Density: 1.195 ‐1.22 MP/Range: ‐30°C Flash Point: 126°C NOTICE: The technical information and suggestions for use made herein are based on VanDeMark’s research and experience and are believed to be reliable, but such information and suggestions do not constitute a warranty, and no patent liability can be assumed. This publication is not to be taken as a license to operate under or infringe on any patents. Since VanDeMark has no control over the conditions under which the product is transported, stored, handled, used or applied, buyer must determine for himself by preliminary tests or otherwise, the suitability of the product for his purposes. VanDeMark’s liability on any basis is limited to the price of the product used. The information in this bulletin supersedes all previously issued bulletins on the subject matter covered. VanDeMark Benzyl Chloroformate APPLICATIONS SPILLS AND DISPOSAL Benzyl Chloroformate is a reactive chemical intermediate Use personal protective equipment (see MSDS). used in the synthesis of pharmaceutical and agrochemical Evacuate personnel to safe areas. Dike far ahead of products. It is used as a reagent in peptide synthesis to liquid spill for later disposal. -

4 Methanesulfonyl Chloride1 Acute Exposure Guideline Levels
Committee on Acute Exposure Guideline Levels Committee on Toxicology Board on Environmental Studies and Toxicology Division on Earth and Life Studies THE NATIONAL ACADEMIES PRESS 500 FIFTH STREET, NW WASHINGTON, DC 20001 NOTICE: The project that is the subject of this report was approved by the Governing Board of the National Research Council, whose members are drawn from the councils of the National Academy of Sciences, the National Academy of Engineering, and the Insti- tute of Medicine. The members of the committee responsible for the report were chosen for their special competences and with regard for appropriate balance. This project was supported by Contract No. W81K04-11-D-0017 and EP-W-09-007 be- tween the National Academy of Sciences and the U.S. Department of Defense and the U.S. Environmental Protection Agency. Any opinions, findings, conclusions, or recom- mendations expressed in this publication are those of the author(s) and do not necessarily reflect the view of the organizations or agencies that provided support for this project. International Standard Book Number-13: 978-0-309-28308-3 International Standard Book Number-10: 0-309-28308-6 Additional copies of this report are available for sale from the National Academies Press, 500 Fifth Street, NW, Keck 360, Washington, DC 20001; (800) 624-6242 or (202) 334- 3313; http://www.nap.edu/. Copyright 2013 by the National Academy of Sciences. All rights reserved. Printed in the United States of America The National Academy of Sciences is a private, nonprofit, self-perpetuating society of distinguished scholars engaged in scientific and engineering research, dedicated to the furtherance of science and technology and to their use for the general welfare.