Studies Towards the Synthesis of the Core Structure of Sarains A, B, and C
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Methanesulfonyl Chloride
A Publication of Reliable Methods for the Preparation of Organic Compounds Working with Hazardous Chemicals The procedures in Organic Syntheses are intended for use only by persons with proper training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at http://www.nap.edu/catalog.php?record_id=12654). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices. In some articles in Organic Syntheses, chemical-specific hazards are highlighted in red “Caution Notes” within a procedure. It is important to recognize that the absence of a caution note does not imply that no significant hazards are associated with the chemicals involved in that procedure. Prior to performing a reaction, a thorough risk assessment should be carried out that includes a review of the potential hazards associated with each chemical and experimental operation on the scale that is planned for the procedure. Guidelines for carrying out a risk assessment and for analyzing the hazards associated with chemicals can be found in Chapter 4 of Prudent Practices. The procedures described in Organic Syntheses are provided as published and are conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein. -

덴드리틱 벤질 클로라이드의 효율적인 합성 Facile Synthesis of Dendritic Benzyl Chlorides from Their Alcohols W
Polymer(Korea), Vol. 31, No. 5, pp 417-421, 2007 덴드리틱 벤질 클로라이드의 효율적인 합성 이재욱F,7ㆍ한승철Fㆍ김희주ㆍ김정환ㆍ이언엽ㆍ김병기ㆍ성새름ㆍ강화신ㆍ김지현FFㆍ허도성FFF 동아대학교 화학과, F동아대학교 의생명과학과, FF동국대학교 생명화학공학과, FFF인제대학교 의생명화학과 (2007년 5월 9일 접수, 2007년 6월 29일 채택) Facile Synthesis of Dendritic Benzyl Chlorides from Their Alcohols with Methanesulfonyl Chloride/Et3N Jae Wook Lee*,7, Seung Choul Han*, Hee Joo Kim, Jung Hwan Kim, Un Yup Lee, Byoung-Ki Kim, Sae Reum Sung, Hwa-Shin Kang, Ji Hyeon Kim**, and Do-Sung Huh*** Department of Chemistry, Dong-A University, Hadan-2-dong, Busan 604-714, Korea *Department of Medical Bioscience, Dong-A University, Hadan-2-dong, Busan 604-714, Korea **Department of Chemical & Biochemical Engineering, Dongguk University, Seoul 100-715, Korea ***Department of Biomedicinal Chemistry, Inje University, Gim Hai 621-749, Korea (Received May 9, 2007; Accepted June 29, 2007) 초록: 덴드리틱 벤질 알코올을 트리에틸아민과 메탄술포닐클로라이드와 반응시켜서, 덴드리틱 벤질 클로라이드 의 효율적인 합성이 이루어졌다. 이 반응은 히드록시기의 메실화 반응과 염소화 반응의 2단계 반응으로 이루어지 는데, 중간체의 분리없이 한 반응 용기내에서 반응이 진행되는 경제적인 방법이다. Abstract: A successful rapid synthesis of dendritic benzyl chlorides from dendritic benzyl alcohols using methanesulfonyl chloride/Et3N as activating agents was described. In this method, each dendritic benzyl chloride can be prepared in one pot: no isolation of intermediate mesylated dendrons is required. The key steps in the syntheses of dendritic benzyl chlorides were the mesylation of the hydroxymethyl group followed by the chlorination by in-situ generated triethylammonium chloride. Keywords: benzylic alcohol, chlorination. Introduction vation and coupling steps in high yield. In addition, the coupling step must be very efficient to enable complete Dendrimers represent a novel type of polymeric material reaction. -

Download The
The Final Program The Final Program for this Symposium was modified from that presented in the Book of Abstracts mainly due to the unforeseen incidence of the coronavirus in late January, 2020. We are grateful to the distinguished scientists who were able to take the place of those who were unable to come, and we regretted the absence of the distinguished scientists who had planned until the last hours to be with us. The SCHEDULE that is posted on this web site is the Final Schedule. That which is in the Book of Abstracts is the schedule that was expected to be the final schedule two weeks prior to the event. Strongly Donating 1,2,3-Triazole-Derived Carbenes for Metal-Mediated Redox Catalysis Simone Bertini, Matteo Planchestainer, Francesca Paradisi, Martin Albrecht Department of Chemistry & Biochemistry, University of Bern, CH-3012 Bern, Switzerland [email protected] Triazole-derived N-heterocyclic carbenes have become an attractive addition to the family of NHC ligands, in parts because their versatile and functional group-tolerant synthesis through click- chemistry,1 and in other parts because of their stronger donor properties to transition metals when compared to ubiquitous Arduengo-type carbenes.2. We have been particularly attracted recently by the high robustness of these ligands towards oxidative and reductive conditions, which provides appealing opportunities for challenging redox catalysis. We have exploited these properties for example for developing iridium complexes as highly active and molecular water oxidation catalysts.3 Here we will present our efforts to use triazole-derived carbenes to enable 1st row transition metals as catalytically competent entities. -

Certified by ...--- ,'
[4 + 1] ANNULATION REACTIONS OF (TRIALKYLSILYL)KETENES: SYNTHESIS OF SUBSTITUTED INDANONES AND CYCLOPENTENONES by Christopher P. Davie B.S., Chemistry Boston College, 1999 SUBMITTED TO THE DEPARTMENT OF CHEMISTRY IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY at the MASSACHUSETTS INSTITUTE OF TECHNOLOGY September 2005 © 2005 Massachusetts Institute of Technology All Rights Reserved Signaure of Author....................... ......... epartmentof Chem..............................istry Department of Chemistry June 7, 2005 Certifiedby ........ ....--- ,'................................ ................. Rick L. Danheiser Thesis Supervisor Acceptedby.................................................................................................... Robert W. Field -- MASSACHUSETTS INSTIUTE Departmental committee on Uraduate Studies I OF TECHNOLOGY OCT 1 12005 ARCHIVEs. LIBRARIES This doctoral thesis has been examined by a committee of the Department of Chemistry as follows: Professor Gregory C. Fu I... ..............; Chairman ]ProfessorProfessor RickRick LL. Danheiser....~cDanheiser ...... ....... ............. .. .................................... Thesis Supervisor Professor Timothy F. Jamison ...... ..... 2 Acknowledgments I would first like to thank my advisor, Rick Danheiser, for his mentoring and support during the past five years. I greatly appreciate Rick's dedication to education, and all that I have learned from him and by working in his lab. I am grateful to Professors Greg Fu and Tim Jamison, members of my thesis committee, for their constructive feedback on this thesis as well as their assistance and advice during my job search. I must also thank Professor Fu for providing valuable advice during our annual meetings. I owe a debt of gratitude to the past and present members of the Danheiser group that I have had the pleasure of working with during my time at MIT. While chemistry was frustrating at times, days in lab were (almost) always enjoyable because of the great group of people working in the lab. -
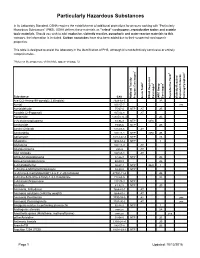
Particularly Hazardous Substances
Particularly Hazardous Substances In its Laboratory Standard, OSHA requires the establishment of additional protections for persons working with "Particularly Hazardous Substances" (PHS). OSHA defines these materials as "select" carcinogens, reproductive toxins and acutely toxic materials. Should you wish to add: explosive, violently reactive, pyrophoric and water-reactve materials to this category, the information is included. Carbon nanotubes have also been added due to their suspected carcinogenic properties. This table is designed to assist the laboratory in the identification of PHS, although it is not definitively conclusive or entirely comprehensive. *Notes on the proper use of this table appear on page 12. 1 6 5 2 3 4 Substance CAS National Toxicity National Program Carcinogen Toxin Acute Regulated OSHA Carcinogen Group IARC Carcinogen Toxin Reproductive Violently Reactive/ Explosive/Peroxide Forming/Pyrophoric A-a-C(2-Amino-9H-pyrido[2,3,b]indole) 2648-68-5 2B Acetal 105-57-7 yes Acetaldehyde 75-07-0 NTP AT 2B Acrolein (2-Propenal) 107-02-8 AT Acetamide 126850-14-4 2B 2-Acetylaminofluorene 53-96-3 NTP ORC Acrylamide 79-06-6 NTP 2B Acrylyl Chloride 814-68-6 AT Acrylonitrile 107-13-1 NTP ORC 2B Adriamycin 23214-92-8 NTP 2A Aflatoxins 1402-68-2 NTP 1 Allylamine 107-11-9 AT Alkylaluminums varies AT Allyl Chloride 107-05-1 AT ortho-Aminoazotoluene 97-56-3 NTP 2B para-aminoazobenzene 60-09-3 2B 4-Aminobiphenyl 92-67-1 NTP ORC 1 1-Amino-2-Methylanthraquinone 82-28-0 NTP (2-Amino-6-methyldipyrido[1,2-a:3’,2’-d]imidazole) 67730-11-4 2B -

Ketenes 25/01/2014 Part 1
Baran Group Meeting Hai Dao Ketenes 25/01/2014 Part 1. Introduction Ph Ph n H Pr3N C A brief history Cl C Ph + nPr NHCl Ph O 3 1828: Synthesis of urea = the starting point of modern organic chemistry. O 1901: Wedekind's proposal for the formation of ketene equivalent (confirmed by Staudinger 1911) Wedekind's proposal (1901) 1902: Wolff rearrangement, Wolff, L. Liebigs Ann. Chem. 1902, 325, 129. 2 Wolff adopt a ketene structure in 1912. R 2 hν R R2 1905: First synthesis and characterization of a ketene: in an efford to synthesize radical 2, 1 ROH R C Staudinger has synthesized diphenylketene 3, Staudinger, H. et al., Chem. Ber. 1905, 1735. N2 1 RO CH or Δ C R C R1 1907-8: synthesis and dicussion about structure of the parent ketene, Wilsmore, O O J. Am. Chem. Soc. 1907, 1938; Wilsmore and Stewart Chem. Ber. 1908, 1025; Staudinger and Wolff rearrangement (1902) O Klever Chem. Ber. 1908, 1516. Ph Ph Cl Zn Ph O hot Pt wire Zn Br Cl Cl CH CH2 Ph C C vs. C Br C Ph Ph HO O O O O O O O 1 3 (isolated) 2 Wilsmore's synthesis and proposal (1907-8) Staudinger's synthesis and proposal (1908) wanted to make Staudinger's discovery (1905) Latest books: ketene (Tidwell, 1995), ketene II (Tidwell, 2006), Science of Synthesis, Vol. 23 (2006); Latest review: new direactions in ketene chemistry: the land of opportunity (Tidwell et al., Eur. J. Org. Chem. 2012, 1081). Search for ketenes, Google gave 406,000 (vs. -

Fragment-Based Discovery of a New Class of Inhibitors Targeting Mycobacterial Trna Modification
Supplementary Data Fragment-based discovery of a new class of inhibitors targeting mycobacterial tRNA modification Sherine E. Thomas*1, Andrew J. Whitehouse*2, Karen Brown3,4, Juan M. Belardinelli5, Ramanuj Lahiri6, M. Daben J. Libardo7, Pooja Gupta1, Sony Malhotra8, Helena I. M. Boshoff7, Mary Jackson5, Chris Abell 2, Anthony G. Coyne2, Tom L. Blundell §1, R. Andres Floto3,4, Vitor Mendes §1 1 Department of Biochemistry, University of Cambridge, 80 Tennis Court Road, Cambridge CB2 1GA, UK. 2 Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge, CB2 1EW, UK. 3 MRC Laboratory of Molecular Biology, Francis Crick Avenue, Cambridge, CB2 0QH, U.K. 4 Cambridge Centre for Lung Infection, Royal Papworth Hospital, Cambridge, CB23 3RE, U.K. 5 Mycobacteria Research Laboratories, Department of Microbiology, Immunology and Pathology, Colorado State University, Fort Collins, Colorado, USA. 6 National Hansen’s Disease Program, Healthcare Systems Bureau, HRSA, DHHS, USA 7 Tuberculosis Research Section, Laboratory of Clinical Immunology and Microbiology, National Institute of Allergy and Infectious Disease, National Institutes of Health, 9000 Rockville Pike, Bethesda, Maryland 20892, USA. 8 Birkbeck College, University of London, Malet Street, WC1E7HX, UK * Contributed equally § To whom correspondence should be addressed Results Conservation of catalytic residues in mycobacterial TrmD A multiple sequence alignment of 5 mycobacterial TrmD enzyme sequences with 10 TrmD ortholog amino acid sequences (Figure S1) shows conservation of important catalytic residues and residues involved in tRNA recognition and methyl transfer reactions. The catalytic residues Asp169 and Arg154 are highly conserved across TrmD orthologs. Key residues such as Asp50, Gly59, His46 involved in tRNA G36 and G37 base recognition, interactions with bases at positions 38, 32 and anticodon branch of wild type tRNA respectively, are also broadly conserved. -

4 Methanesulfonyl Chloride1 Acute Exposure Guideline Levels
Committee on Acute Exposure Guideline Levels Committee on Toxicology Board on Environmental Studies and Toxicology Division on Earth and Life Studies THE NATIONAL ACADEMIES PRESS 500 FIFTH STREET, NW WASHINGTON, DC 20001 NOTICE: The project that is the subject of this report was approved by the Governing Board of the National Research Council, whose members are drawn from the councils of the National Academy of Sciences, the National Academy of Engineering, and the Insti- tute of Medicine. The members of the committee responsible for the report were chosen for their special competences and with regard for appropriate balance. This project was supported by Contract No. W81K04-11-D-0017 and EP-W-09-007 be- tween the National Academy of Sciences and the U.S. Department of Defense and the U.S. Environmental Protection Agency. Any opinions, findings, conclusions, or recom- mendations expressed in this publication are those of the author(s) and do not necessarily reflect the view of the organizations or agencies that provided support for this project. International Standard Book Number-13: 978-0-309-28308-3 International Standard Book Number-10: 0-309-28308-6 Additional copies of this report are available for sale from the National Academies Press, 500 Fifth Street, NW, Keck 360, Washington, DC 20001; (800) 624-6242 or (202) 334- 3313; http://www.nap.edu/. Copyright 2013 by the National Academy of Sciences. All rights reserved. Printed in the United States of America The National Academy of Sciences is a private, nonprofit, self-perpetuating society of distinguished scholars engaged in scientific and engineering research, dedicated to the furtherance of science and technology and to their use for the general welfare. -

Guide for the Selection of Chemical and Biological Decontamination Equipment for Emergency First Responders
U.S. Department of Justice Office of Justice Programs National Institute of Justice National Institute of Justice Law Enforcement and Corrections Standards and Testing Program Guide for the Selection of Chemical and Biological Decontamination Equipment for Emergency First Responders NIJ Guide 103–00 Volume I October 2001 ABOUT THE LAW ENFORCEMENT AND CORRECTIONS STANDARDS AND TESTING PROGRAM The Law Enforcement and Corrections Standards and Testing Program is sponsored by the Office of Science and Technology of the National Institute of Justice (NIJ), U.S. Department of Justice. The program responds to the mandate of the Justice System Improvement Act of 1979, which directed NIJ to encourage research and development to improve the criminal justice system and to disseminate the results to Federal, State, and local agencies. The Law Enforcement and Corrections Standards and Testing Program is an applied research effort that determines the technological needs of justice system agencies, sets minimum performance standards for specific devices, tests commercially available equipment against those standards, and disseminates the standards and the test results to criminal justice agencies nationally and internationally. The program operates through: The Law Enforcement and Corrections Technology Advisory Council (LECTAC), consisting of nationally recognized criminal justice practitioners from Federal, State, and local agencies, which assesses technological needs and sets priorities for research programs and items to be evaluated and tested. The Office of Law Enforcement Standards (OLES) at the National Institute of Standards and Technology, which develops voluntary national performance standards for compliance testing to ensure that individual items of equipment are suitable for use by criminal justice agencies. -

Carbene Rearrangements: Intramolecular Interaction of a Triple Bond with a Carbene Center
An Abstract OF THE THESIS OF Jose C. Danino for the degree of Doctor of Philosophy in Chemistry presented on _Dcc, Title: Carbene RearrangementE) Intramolecular Interaction of a Triple Bond with aCarbene Center Redacted for Privacy Abstract approved: Dr. Vetere. Freeman The tosylhydrazones of2-heptanone, 4,4-dimethy1-2- heptanone, 6-heptyn-2-one and 4,4-dimethy1-6-heptyn-2- one were synthesizedand decomposed under a varietyof reaction conditions:' drylithium and sodium salt pyrolyses, sodium methoxide thermolysesin diglyme and photolyses of the lithium salt intetrahydrofuran. The saturated ana- logues 2-heptanone tosylhydrazoneand its 4,4-dimethyl isomer afforded the alkenesarising from 6-hydrogeninser- product distribution in the tion. It was determined that differ- dry salt pyrolyses of2-heptanone tosylhydrazone was ent for the lithiumand the sodium salts. However, the product distribution of thedry sodium salt was verysimilar diglyme to product distributionobtained on thermolysis in explained by a with sodium methoxide. This difference was reaction of lithium bromide(present as an impurity inall compound to the lithium salts)with the intermediate diazo afford an organolithiumintermediate that behaves in a some- what different fashionthan the free carbene.The unsaturated analogues were found to produce a cyclic product in addition to the expected acyclic alkenes arising from 3-hydrogen insertion. By comparison of the acyclic alkene distri- bution obtained in the saturated analogues with those in the unsaturated analogues, it was concluded that at leastsome cyclization was occurring via addition of the diazo moiety to the triple bond. It was determined that the organo- lithium intermediateresulting from lithium bromide cat- alyzed decomposition of the diazo compound was incapable of cyclization. -

Guide for the Selection of Personal Protective Equipment for Emergency First Responders
U.S. Department of Justice Office of Justice Programs National Institute of Justice National Institute of Justice Law Enforcement and Corrections Standards and Testing Program Guide for the Selection of Personal Protective Equipment for Emergency First Responders NIJ Guide 102–00 Volume I November 2002 U.S. Department of Justice Office of Justice Programs 810 Seventh Street N.W. Washington, DC 20531 John Ashcroft Attorney General Deborah J. Daniels Assistant Attorney General Sarah V. Hart Director, National Institute of Justice For grant and funding information, contact: Department of Justice Response Center 800–421–6770 Office of Justice Programs National Institute of Justice World Wide Web Site World Wide Web Site http://www.ojp.usdoj.gov http://www.ojp.usdoj.gov/nij U.S. Department of Justice Office of Justice Programs National Institute of Justice Guide for the Selection of Personal Protective Equipment for Emergency First Responders NIJ Guide 102-00, Volume I Dr. Alim A. Fatah1 John A. Barrett2 Richard D. Arcilesi, Jr.2 Charlotte H. Lattin2 Charles G. Janney2 Edward A. Blackman2 Coordination by: Office of Law Enforcement Standards National Institute of Standards and Technology Gaithersburg, MD 20899–8102 Prepared for: National Institute of Justice Office of Science and Technology Washington, DC 20531 November 2002 This document was prepared under CBIAC contract number SPO-900-94-D-0002 and Interagency Agreement M92361 between NIST and the Department of Defense Technical Information Center (DTIC). NCJ 191518 1National Institute of Standards and Technology, Office of Law Enforcement Standards. 2Battelle Memorial Institute. National Institute of Justice Sarah V. Hart Director This guide was prepared for the National Institute of Justice, U.S. -

Reactions of (Trialkylsilyl)Vinylketenes with Lithium Ynolates: a New Benzannulation Strategy Wesley F. Austin, Yongjung Zhang
Reactions of (Trialkylsilyl)vinylketenes with Lithium Ynolates: A New Benzannulation Strategy Wesley F. Austin, Yongjung Zhang, and Rick L. Danheiser* Department of Chemistry Massachusetts Institute of Technology Cambridge, Massachusetts 02139 Supporting Information General Procedures. All reactions were performed in flame-dried or oven-dried glassware under a positive pressure of argon. Reaction mixtures were stirred magnetically unless otherwise indicated. Air- and moisture-sensitive liquids and solutions were transferred by syringe or cannula and were introduced into reaction vessels through rubber septa. Reaction product solutions and chromatography fractions were concentrated by rotary evaporation at ca. 20 mmHg and then at ca. 0.1 mmHg (vacuum pump) unless otherwise indicated. Thin layer chromatography was performed on Merck precoated glass- backed silica gel 60 F-254 0.25 mm plates. Column chromatography was performed on Silicycle silica gel 60 (230-400 mesh), or Sorbent silica gel 60A (32-63 µm). Materials. (Triisopropylsilyl)vinylketenes 6a and 6b were prepared as described previously.1 2- Cyclohexyl-1-(triisopropylsiloxy)ethyne (9a) and 3-methyl-1-(triisopropylsiloxy)-1-butyne (9b) were prepared from ethyl cyclohexanecarboxylate2 and ethyl isobutyrate3 respectively via the one-step Kowalski reaction as reported previously. Siloxy alkynes 9d4 and 9e5 have been prepared from the corresponding alkynes by employing the method of Julia.6 Anhydrous tert-butyl hydroperoxide in toluene was prepared from the aqueous solution as described by Sharpless.7 Commercial grade reagents and solvents were used without further purification except as indicated below. Triisopropylsilyl trifluoromethanesulfonate, 1,1,1,3,3,3-hexamethyldisilazane, hexanes, benzene, and 1 Loebach, J. L.; Bennett, D. M.; Danheiser, R.