1078-0432.CCR-10-0889.Full.Pdf
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Investigation of Candidate Genes and Mechanisms Underlying Obesity
Prashanth et al. BMC Endocrine Disorders (2021) 21:80 https://doi.org/10.1186/s12902-021-00718-5 RESEARCH ARTICLE Open Access Investigation of candidate genes and mechanisms underlying obesity associated type 2 diabetes mellitus using bioinformatics analysis and screening of small drug molecules G. Prashanth1 , Basavaraj Vastrad2 , Anandkumar Tengli3 , Chanabasayya Vastrad4* and Iranna Kotturshetti5 Abstract Background: Obesity associated type 2 diabetes mellitus is a metabolic disorder ; however, the etiology of obesity associated type 2 diabetes mellitus remains largely unknown. There is an urgent need to further broaden the understanding of the molecular mechanism associated in obesity associated type 2 diabetes mellitus. Methods: To screen the differentially expressed genes (DEGs) that might play essential roles in obesity associated type 2 diabetes mellitus, the publicly available expression profiling by high throughput sequencing data (GSE143319) was downloaded and screened for DEGs. Then, Gene Ontology (GO) and REACTOME pathway enrichment analysis were performed. The protein - protein interaction network, miRNA - target genes regulatory network and TF-target gene regulatory network were constructed and analyzed for identification of hub and target genes. The hub genes were validated by receiver operating characteristic (ROC) curve analysis and RT- PCR analysis. Finally, a molecular docking study was performed on over expressed proteins to predict the target small drug molecules. Results: A total of 820 DEGs were identified between -

Role and Regulation of the P53-Homolog P73 in the Transformation of Normal Human Fibroblasts
Role and regulation of the p53-homolog p73 in the transformation of normal human fibroblasts Dissertation zur Erlangung des naturwissenschaftlichen Doktorgrades der Bayerischen Julius-Maximilians-Universität Würzburg vorgelegt von Lars Hofmann aus Aschaffenburg Würzburg 2007 Eingereicht am Mitglieder der Promotionskommission: Vorsitzender: Prof. Dr. Dr. Martin J. Müller Gutachter: Prof. Dr. Michael P. Schön Gutachter : Prof. Dr. Georg Krohne Tag des Promotionskolloquiums: Doktorurkunde ausgehändigt am Erklärung Hiermit erkläre ich, dass ich die vorliegende Arbeit selbständig angefertigt und keine anderen als die angegebenen Hilfsmittel und Quellen verwendet habe. Diese Arbeit wurde weder in gleicher noch in ähnlicher Form in einem anderen Prüfungsverfahren vorgelegt. Ich habe früher, außer den mit dem Zulassungsgesuch urkundlichen Graden, keine weiteren akademischen Grade erworben und zu erwerben gesucht. Würzburg, Lars Hofmann Content SUMMARY ................................................................................................................ IV ZUSAMMENFASSUNG ............................................................................................. V 1. INTRODUCTION ................................................................................................. 1 1.1. Molecular basics of cancer .......................................................................................... 1 1.2. Early research on tumorigenesis ................................................................................. 3 1.3. Developing -
Drosophila and Human Transcriptomic Data Mining Provides Evidence for Therapeutic
Drosophila and human transcriptomic data mining provides evidence for therapeutic mechanism of pentylenetetrazole in Down syndrome Author Abhay Sharma Institute of Genomics and Integrative Biology Council of Scientific and Industrial Research Delhi University Campus, Mall Road Delhi 110007, India Tel: +91-11-27666156, Fax: +91-11-27662407 Email: [email protected] Nature Precedings : hdl:10101/npre.2010.4330.1 Posted 5 Apr 2010 Running head: Pentylenetetrazole mechanism in Down syndrome 1 Abstract Pentylenetetrazole (PTZ) has recently been found to ameliorate cognitive impairment in rodent models of Down syndrome (DS). The mechanism underlying PTZ’s therapeutic effect is however not clear. Microarray profiling has previously reported differential expression of genes in DS. No mammalian transcriptomic data on PTZ treatment however exists. Nevertheless, a Drosophila model inspired by rodent models of PTZ induced kindling plasticity has recently been described. Microarray profiling has shown PTZ’s downregulatory effect on gene expression in fly heads. In a comparative transcriptomics approach, I have analyzed the available microarray data in order to identify potential mechanism of PTZ action in DS. I find that transcriptomic correlates of chronic PTZ in Drosophila and DS counteract each other. A significant enrichment is observed between PTZ downregulated and DS upregulated genes, and a significant depletion between PTZ downregulated and DS dowwnregulated genes. Further, the common genes in PTZ Nature Precedings : hdl:10101/npre.2010.4330.1 Posted 5 Apr 2010 downregulated and DS upregulated sets show enrichment for MAP kinase pathway. My analysis suggests that downregulation of MAP kinase pathway may mediate therapeutic effect of PTZ in DS. Existing evidence implicating MAP kinase pathway in DS supports this observation. -

Robust Identification of Local Adaptation from Allele
INVESTIGATION Robust Identification of Local Adaptation from Allele Frequencies Torsten Günther*,1 and Graham Coop†,1 *Institute of Plant Breeding, Seed Science, and Population Genetics, University of Hohenheim, 70593 Stuttgart, Germany, and †Department of Evolution and Ecology and Center for Population Biology, University of California, Davis, California 95616 ABSTRACT Comparing allele frequencies among populations that differ in environment has long been a tool for detecting loci involved in local adaptation. However, such analyses are complicated by an imperfect knowledge of population allele frequencies and neutral correlations of allele frequencies among populations due to shared population history and gene flow. Here we develop a set of methods to robustly test for unusual allele frequency patterns and correlations between environmental variables and allele frequencies while accounting for these complications based on a Bayesian model previously implemented in the software Bayenv. Using this model, we calculate a set of “standardized allele frequencies” that allows investigators to apply tests of their choice to multiple populations while accounting for sampling and covariance due to population history. We illustrate this first by showing that these standardized frequencies can be used to detect nonparametric correlations with environmental variables; these correlations are also less prone to spurious results due to outlier populations. We then demonstrate how these standardized allele frequencies can be used to construct a test to detect SNPs that deviate strongly from neutral population structure. This test is conceptually related to FST and is shown to be more powerful, as we account for population history. We also extend the model to next-generation sequencing of population pools— a cost-efficient way to estimate population allele frequencies, but one that introduces an additional level of sampling noise. -

RABIF/MSS4 Is a Rab-Stabilizing Holdase Chaperone Required for GLUT4 Exocytosis
RABIF/MSS4 is a Rab-stabilizing holdase chaperone required for GLUT4 exocytosis Daniel R. Gulbransona, Eric M. Davisa, Brittany A. Demmitta,b, Yan Ouyanga, Yihong Yec, Haijia Yua,d,1, and Jingshi Shena,1 aDepartment of Molecular, Cellular and Developmental Biology, University of Colorado, Boulder, CO 80309; bInstitute for Behavioral Genetics, University of Colorado, Boulder, CO 80309; cLaboratory of Molecular Biology, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, MD 20892; and dJiangsu Key Laboratory for Molecular and Medical Biotechnology, College of Life Sciences, Nanjing Normal University, Nanjing 210023, China Edited by Jennifer Lippincott-Schwartz, Howard Hughes Medical Institute, Ashburn, VA, and approved August 21, 2017 (received for review July 7, 2017) Rab GTPases are switched from their GDP-bound inactive confor- anabolic hormone insulin facilitates glucose uptake by acutely mation to a GTP-bound active state by guanine nucleotide exchange relocating GLUT4 from intracellular compartments to the cell factors (GEFs). The first putative GEFs isolated for Rabs are RABIF surface (6, 20, 21, 23). Upon the termination of insulin signaling, (Rab-interacting factor)/MSS4 (mammalian suppressor of Sec4) and GLUT4 is retrieved from the plasma membrane through endocy- its yeast homolog DSS4 (dominant suppressor of Sec4). However, tosis and returns to intracellular storage vesicles (6). Importantly, the biological function and molecular mechanism of these molecules the components of GLUT4 exocytosis are also involved in the remained unclear. In a genome-wide CRISPR genetic screen, we regulation of other exocytic pathways such as insulin secretion and isolated RABIF as a positive regulator of exocytosis. -

WO 2014/205555 Al 31 December 2014 (31.12.2014) P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2014/205555 Al 31 December 2014 (31.12.2014) P O P C T (51) International Patent Classification: British Columbia V5Y 2G1 (CA). WATAHIKI, Akira; A61K 31/713 (2006.01) G01N 33/48 (2006.01) 15-5 Takakura-cho, Nishinomiya, 62-0872 (JP). LIU, Hui A61K 31/7088 (2006.01) G01N 33/50 (2006.01) Hsuan; 207 - 5770 Oak Street, Vancouver, British A61P 35/00 (2006.01) CI2N 15/113 (2010.01) Columbia V6M 4M5 (CA). PAROLIA, Abhijit; 505 C12Q 1/68 (2006.01) Mumfordganj, 2 11002 Allahabad U.P. (IN). (21) International Application Number: (74) Agents: SECHLEY, Konrad et al; Gowling Lafleur PCT/CA2014/000538 Henderson LLP, 2300, 550 Burrard Street, Vancouver, British Columbia V6C 2B5 (CA). (22) International Filing Date: 27 June 2014 (27.06.2014) (81) Designated States (unless otherwise indicated, for every kind of national protection available): AE, AG, AL, AM, (25) Filing Language: English AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, (26) Publication Language: English BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, (30) Priority Data: HN, HR, HU, ID, IL, IN, IR, IS, JP, KE, KG, KN, KP, KR, 61/841,225 28 June 2013 (28.06.2013) US KZ, LA, LC, LK, LR, LS, LT, LU, LY, MA, MD, ME, (71) Applicant: BRITISH COLUMBIA CANCER AGENCY MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, BRANCH [CA/CA]; 675 West 10th Avenue, Vancouver, OM, PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, British Columbia V5Z 1L3 (CA). -

Promoterless Transposon Mutagenesis Drives Solid Cancers Via Tumor Suppressor Inactivation
bioRxiv preprint doi: https://doi.org/10.1101/2020.08.17.254565; this version posted August 17, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 Promoterless Transposon Mutagenesis Drives Solid Cancers via Tumor Suppressor Inactivation 2 Aziz Aiderus1, Ana M. Contreras-Sandoval1, Amanda L. Meshey1, Justin Y. Newberg1,2, Jerrold M. Ward3, 3 Deborah Swing4, Neal G. Copeland2,3,4, Nancy A. Jenkins2,3,4, Karen M. Mann1,2,3,4,5,6,7, and Michael B. 4 Mann1,2,3,4,6,7,8,9 5 1Department of Molecular Oncology, Moffitt Cancer Center & Research Institute, Tampa, FL, USA 6 2Cancer Research Program, Houston Methodist Research Institute, Houston, Texas, USA 7 3Institute of Molecular and Cell Biology, Agency for Science, Technology and Research (A*STAR), 8 Singapore, Republic of Singapore 9 4Mouse Cancer Genetics Program, Center for Cancer Research, National Cancer Institute, Frederick, 10 Maryland, USA 11 5Departments of Gastrointestinal Oncology & Malignant Hematology, Moffitt Cancer Center & Research 12 Institute, Tampa, FL, USA 13 6Cancer Biology and Evolution Program, Moffitt Cancer Center & Research Institute, Tampa, FL, USA 14 7Department of Oncologic Sciences, Morsani College of Medicine, University of South Florida, Tampa, FL, 15 USA. 16 8Donald A. Adam Melanoma and Skin Cancer Research Center of Excellence, Moffitt Cancer Center, Tampa, 17 FL, USA 18 9Department of Cutaneous Oncology, Moffitt Cancer Center & Research Institute, Tampa, FL, USA 19 These authors contributed equally: Aziz Aiderus, Ana M. -
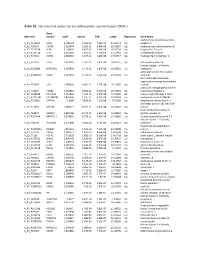
Table S1. the Statistical Metrics for Key Differentially Expressed Genes (Degs)
Table S1. The statistical metrics for key differentially expressed genes (DEGs) Gene Agilent Id Symbol logFC pValue FDR tvalue Regulation Gene Name oxidized low density lipoprotein A_24_P124624 OLR1 2.458429 1.19E-13 7.25E-10 24.04241 Up receptor 1 A_23_P90273 CHST8 2.622464 3.85E-12 6.96E-09 19.05867 Up carbohydrate sulfotransferase 8 A_23_P217528 KLF8 2.109007 4.85E-12 7.64E-09 18.76234 Up Kruppel like factor 8 A_23_P114740 CFH 2.651636 1.85E-11 1.79E-08 17.13652 Up complement factor H A_23_P34031 XAGE2 2.000935 2.04E-11 1.81E-08 17.02457 Up X antigen family member 2 A_23_P27332 TCF4 1.613097 2.32E-11 1.87E-08 16.87275 Up transcription factor 4 histone cluster 1 H1 family A_23_P250385 HIST1H1B 2.298658 2.47E-11 1.87E-08 16.80362 Up member b abnormal spindle microtubule A_33_P3288159 ASPM 2.162032 2.79E-11 2.01E-08 16.66292 Up assembly H19, imprinted maternally expressed transcript (non-protein A_24_P52697 H19 1.499364 4.09E-11 2.76E-08 16.23387 Up coding) potassium voltage-gated channel A_24_P31627 KCNB1 2.289689 6.65E-11 3.97E-08 15.70253 Up subfamily B member 1 A_23_P214168 COL12A1 2.155835 7.59E-11 4.15E-08 15.56005 Up collagen type XII alpha 1 chain A_33_P3271341 LOC388282 2.859496 7.61E-11 4.15E-08 15.55704 Up uncharacterized LOC388282 A_32_P150891 DIAPH3 2.2068 7.83E-11 4.22E-08 15.5268 Up diaphanous related formin 3 zinc finger protein 185 with LIM A_23_P11025 ZNF185 1.385721 8.74E-11 4.59E-08 15.41041 Up domain heat shock protein family B A_23_P96872 HSPB11 1.887166 8.94E-11 4.64E-08 15.38599 Up (small) member 11 A_23_P107454 -

Coexpression Networks Based on Natural Variation in Human Gene Expression at Baseline and Under Stress
University of Pennsylvania ScholarlyCommons Publicly Accessible Penn Dissertations Fall 2010 Coexpression Networks Based on Natural Variation in Human Gene Expression at Baseline and Under Stress Renuka Nayak University of Pennsylvania, [email protected] Follow this and additional works at: https://repository.upenn.edu/edissertations Part of the Computational Biology Commons, and the Genomics Commons Recommended Citation Nayak, Renuka, "Coexpression Networks Based on Natural Variation in Human Gene Expression at Baseline and Under Stress" (2010). Publicly Accessible Penn Dissertations. 1559. https://repository.upenn.edu/edissertations/1559 This paper is posted at ScholarlyCommons. https://repository.upenn.edu/edissertations/1559 For more information, please contact [email protected]. Coexpression Networks Based on Natural Variation in Human Gene Expression at Baseline and Under Stress Abstract Genes interact in networks to orchestrate cellular processes. Here, we used coexpression networks based on natural variation in gene expression to study the functions and interactions of human genes. We asked how these networks change in response to stress. First, we studied human coexpression networks at baseline. We constructed networks by identifying correlations in expression levels of 8.9 million gene pairs in immortalized B cells from 295 individuals comprising three independent samples. The resulting networks allowed us to infer interactions between biological processes. We used the network to predict the functions of poorly-characterized human genes, and provided some experimental support. Examining genes implicated in disease, we found that IFIH1, a diabetes susceptibility gene, interacts with YES1, which affects glucose transport. Genes predisposing to the same diseases are clustered non-randomly in the network, suggesting that the network may be used to identify candidate genes that influence disease susceptibility. -

Patterns of Sequence Conservation in Presynaptic Neural Genes
University of Pennsylvania ScholarlyCommons Departmental Papers (CIS) Department of Computer & Information Science November 2006 Patterns of Sequence Conservation in Presynaptic Neural Genes Dexter Hadley University of Pennsylvania Tara Murphy University of Pennsylvania Otto Valladares University of Pennsylvania Sridhar Hannenhalli University of Pennsylvania Lyle H. Ungar University of Pennsylvania, [email protected] See next page for additional authors Follow this and additional works at: https://repository.upenn.edu/cis_papers Recommended Citation Dexter Hadley, Tara Murphy, Otto Valladares, Sridhar Hannenhalli, Lyle H. Ungar, Junhyong Kim, and Maja Bucan, "Patterns of Sequence Conservation in Presynaptic Neural Genes", . November 2006. Reprinted from Genome Biology, Volume 7, Issue 11, November 2006, pages R105.1-R105.19. Publisher URL: http://genomebiology.com/2006/7/11/R105 This paper is posted at ScholarlyCommons. https://repository.upenn.edu/cis_papers/282 For more information, please contact [email protected]. Patterns of Sequence Conservation in Presynaptic Neural Genes Abstract Background: The neuronal synapse is a fundamental functional unit in the central nervous system of animals. Because synaptic function is evolutionarily conserved, we reasoned that functional sequences of genes and related genomic elements known to play important roles in neurotransmitter release would also be conserved. Results: Evolutionary rate analysis revealed that presynaptic proteins evolve slowly, although some members of large gene families exhibit accelerated evolutionary rates relative to other family members. Comparative sequence analysis of 46 megabases spanning 150 presynaptic genes identified more than 26,000 elements that are highly conserved in eight vertebrate species, as well as a small subset of sequences (6%) that are shared among unrelated presynaptic genes. -

Table S1. 103 Ferroptosis-Related Genes Retrieved from the Genecards
Table S1. 103 ferroptosis-related genes retrieved from the GeneCards. Gene Symbol Description Category GPX4 Glutathione Peroxidase 4 Protein Coding AIFM2 Apoptosis Inducing Factor Mitochondria Associated 2 Protein Coding TP53 Tumor Protein P53 Protein Coding ACSL4 Acyl-CoA Synthetase Long Chain Family Member 4 Protein Coding SLC7A11 Solute Carrier Family 7 Member 11 Protein Coding VDAC2 Voltage Dependent Anion Channel 2 Protein Coding VDAC3 Voltage Dependent Anion Channel 3 Protein Coding ATG5 Autophagy Related 5 Protein Coding ATG7 Autophagy Related 7 Protein Coding NCOA4 Nuclear Receptor Coactivator 4 Protein Coding HMOX1 Heme Oxygenase 1 Protein Coding SLC3A2 Solute Carrier Family 3 Member 2 Protein Coding ALOX15 Arachidonate 15-Lipoxygenase Protein Coding BECN1 Beclin 1 Protein Coding PRKAA1 Protein Kinase AMP-Activated Catalytic Subunit Alpha 1 Protein Coding SAT1 Spermidine/Spermine N1-Acetyltransferase 1 Protein Coding NF2 Neurofibromin 2 Protein Coding YAP1 Yes1 Associated Transcriptional Regulator Protein Coding FTH1 Ferritin Heavy Chain 1 Protein Coding TF Transferrin Protein Coding TFRC Transferrin Receptor Protein Coding FTL Ferritin Light Chain Protein Coding CYBB Cytochrome B-245 Beta Chain Protein Coding GSS Glutathione Synthetase Protein Coding CP Ceruloplasmin Protein Coding PRNP Prion Protein Protein Coding SLC11A2 Solute Carrier Family 11 Member 2 Protein Coding SLC40A1 Solute Carrier Family 40 Member 1 Protein Coding STEAP3 STEAP3 Metalloreductase Protein Coding ACSL1 Acyl-CoA Synthetase Long Chain Family Member 1 Protein -

Transcriptional Features of Multiple Myeloma Patients with Chromosome 1Q Gain
Letters to the Editor 1113 References 5 Dworzak MN, Froschl G, Printz D, Mann G, Po¨tschger U, Mu¨hlegger N et al. Prognostic significance and modalities of flow cytometric minimal residual disease detection in childhood acute 1 Woessmann W, Seidemann K, Mann G, Zimmermann M, Burkhardt lymphoblastic leukemia. Blood 2002; 99: 1952–1958. B, Oschlies I et al. The impact of the methotrexate administration 6 Hummel M, Bentink S, Berger H, Klapper W, Wessendorf S, Barth schedule and dose in the treatment of children and adolescents with TF et al. A biologic definition of Burkitt’s lymphoma from B-cell neoplasms: a report of the BFM Group Study NHL-BFM95. transcriptional and genomic profiling. N Engl J Med 2006; 354: Blood 2005; 105: 948–958. 2419–2430. 2 Busch K, Borkhardt A, Wossmann W, Reiter A, Harbott J. Combined 7 Dave SS, Fu K, Wright GW, Lam LT, Kluin P, Boerma EJ et al. polymerase chain reaction methods to detect c-myc/IgH rearrange- Molecular diagnosis of Burkitt’s lymphoma. N Engl J Med 2006; ment in childhood Burkitt’s lymphoma for minimal residual disease 354: 2431–2442. analysis. Haematologica 2004; 89: 818–825. 8 Attarbaschi A, Mann G, Dworzak M, Trebo M, Urban C, Fink FM 3 Mussolin L, Basso K, Pillon M, d’Amore ES, Lombardi A, Luzzatto L et al. Malignant non-Hodgkin’s lymphoma of childhood and et al. Prospective analysis of minimal bone marrow infiltration in adolescence in Austria – therapy results between 1986 and 2000. pediatric Burkitt’s lymphomas by long-distance polymerase chain Wien Klin Wochenschr 2002; 114: 978–986.