MOF-Associated Complexes Ensure Stem Cell Identity and Xist Repression
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Identification of the Binding Partners for Hspb2 and Cryab Reveals
Brigham Young University BYU ScholarsArchive Theses and Dissertations 2013-12-12 Identification of the Binding arP tners for HspB2 and CryAB Reveals Myofibril and Mitochondrial Protein Interactions and Non- Redundant Roles for Small Heat Shock Proteins Kelsey Murphey Langston Brigham Young University - Provo Follow this and additional works at: https://scholarsarchive.byu.edu/etd Part of the Microbiology Commons BYU ScholarsArchive Citation Langston, Kelsey Murphey, "Identification of the Binding Partners for HspB2 and CryAB Reveals Myofibril and Mitochondrial Protein Interactions and Non-Redundant Roles for Small Heat Shock Proteins" (2013). Theses and Dissertations. 3822. https://scholarsarchive.byu.edu/etd/3822 This Thesis is brought to you for free and open access by BYU ScholarsArchive. It has been accepted for inclusion in Theses and Dissertations by an authorized administrator of BYU ScholarsArchive. For more information, please contact [email protected], [email protected]. Identification of the Binding Partners for HspB2 and CryAB Reveals Myofibril and Mitochondrial Protein Interactions and Non-Redundant Roles for Small Heat Shock Proteins Kelsey Langston A thesis submitted to the faculty of Brigham Young University in partial fulfillment of the requirements for the degree of Master of Science Julianne H. Grose, Chair William R. McCleary Brian Poole Department of Microbiology and Molecular Biology Brigham Young University December 2013 Copyright © 2013 Kelsey Langston All Rights Reserved ABSTRACT Identification of the Binding Partners for HspB2 and CryAB Reveals Myofibril and Mitochondrial Protein Interactors and Non-Redundant Roles for Small Heat Shock Proteins Kelsey Langston Department of Microbiology and Molecular Biology, BYU Master of Science Small Heat Shock Proteins (sHSP) are molecular chaperones that play protective roles in cell survival and have been shown to possess chaperone activity. -

Physical and Functional Interaction Between SET1/COMPASS Complex Component CFP-1 and a Sin3s HDAC Complex in C. Elegans
Physical and functional interaction between SET1/COMPASS complex component CFP-1 and a Sin3S HDAC complex in C. elegans Flore Beurton, Przemyslaw Stempor, Matthieu Caron, Alex Appert, Yan Dong, Ron A-J Chen, David Cluet, Yohann Couté, Marion Herbette, Ni Huang, et al. To cite this version: Flore Beurton, Przemyslaw Stempor, Matthieu Caron, Alex Appert, Yan Dong, et al.. Physical and functional interaction between SET1/COMPASS complex component CFP-1 and a Sin3S HDAC complex in C. elegans. Nucleic Acids Research, Oxford University Press, 2019, 47 (21), pp.11164- 11180. 10.1093/nar/gkz880. hal-02375443 HAL Id: hal-02375443 https://hal.archives-ouvertes.fr/hal-02375443 Submitted on 25 Nov 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. bioRxiv preprint doi: https://doi.org/10.1101/436147; this version posted October 5, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Physical and functional interaction between SET1/COMPASS complex component CFP- 1 and a Sin3 HDAC complex Beurton F. -

Proteomic Identification of the Transcription Factors Ikaros And
European School of Molecular Medicine (SEMM) University of Milan and University of Naples “Federico II” PhD degree in Systems Medicine (curriculum in Molecular Oncology) Settore disciplinare: BIO/11 Proteomic identification of the transcription factors Ikaros and Aiolos as new Myc interactors on chromatin Chiara Veronica Locarno Matricola: R10755 Center for Genomic Science IIT@SEMM, Milan Supervisor: Bruno Amati, PhD IEO, Milan Added Supervisor: Arianna Sabò, PhD IEO, Milan Academic year 2017-2018 Table of contents List of abbreviations ........................................................................................................... 4 List of figures ....................................................................................................................... 8 List of tables ....................................................................................................................... 11 Abstract .............................................................................................................................. 12 1. INTRODUCTION ......................................................................................................... 13 1.1 Myc ........................................................................................................................................ 13 1.1.1 Myc discovery and structure ........................................................................................... 13 1.1.2. Role of Myc in physiological and pathological conditions ........................................... -

Interoperability in Toxicology: Connecting Chemical, Biological, and Complex Disease Data
INTEROPERABILITY IN TOXICOLOGY: CONNECTING CHEMICAL, BIOLOGICAL, AND COMPLEX DISEASE DATA Sean Mackey Watford A dissertation submitted to the faculty at the University of North Carolina at Chapel Hill in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Gillings School of Global Public Health (Environmental Sciences and Engineering). Chapel Hill 2019 Approved by: Rebecca Fry Matt Martin Avram Gold David Reif Ivan Rusyn © 2019 Sean Mackey Watford ALL RIGHTS RESERVED ii ABSTRACT Sean Mackey Watford: Interoperability in Toxicology: Connecting Chemical, Biological, and Complex Disease Data (Under the direction of Rebecca Fry) The current regulatory framework in toXicology is expanding beyond traditional animal toXicity testing to include new approach methodologies (NAMs) like computational models built using rapidly generated dose-response information like US Environmental Protection Agency’s ToXicity Forecaster (ToXCast) and the interagency collaborative ToX21 initiative. These programs have provided new opportunities for research but also introduced challenges in application of this information to current regulatory needs. One such challenge is linking in vitro chemical bioactivity to adverse outcomes like cancer or other complex diseases. To utilize NAMs in prediction of compleX disease, information from traditional and new sources must be interoperable for easy integration. The work presented here describes the development of a bioinformatic tool, a database of traditional toXicity information with improved interoperability, and efforts to use these new tools together to inform prediction of cancer and complex disease. First, a bioinformatic tool was developed to provide a ranked list of Medical Subject Heading (MeSH) to gene associations based on literature support, enabling connection of compleX diseases to genes potentially involved. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

4-6 Weeks Old Female C57BL/6 Mice Obtained from Jackson Labs Were Used for Cell Isolation
Methods Mice: 4-6 weeks old female C57BL/6 mice obtained from Jackson labs were used for cell isolation. Female Foxp3-IRES-GFP reporter mice (1), backcrossed to B6/C57 background for 10 generations, were used for the isolation of naïve CD4 and naïve CD8 cells for the RNAseq experiments. The mice were housed in pathogen-free animal facility in the La Jolla Institute for Allergy and Immunology and were used according to protocols approved by the Institutional Animal Care and use Committee. Preparation of cells: Subsets of thymocytes were isolated by cell sorting as previously described (2), after cell surface staining using CD4 (GK1.5), CD8 (53-6.7), CD3ε (145- 2C11), CD24 (M1/69) (all from Biolegend). DP cells: CD4+CD8 int/hi; CD4 SP cells: CD4CD3 hi, CD24 int/lo; CD8 SP cells: CD8 int/hi CD4 CD3 hi, CD24 int/lo (Fig S2). Peripheral subsets were isolated after pooling spleen and lymph nodes. T cells were enriched by negative isolation using Dynabeads (Dynabeads untouched mouse T cells, 11413D, Invitrogen). After surface staining for CD4 (GK1.5), CD8 (53-6.7), CD62L (MEL-14), CD25 (PC61) and CD44 (IM7), naïve CD4+CD62L hiCD25-CD44lo and naïve CD8+CD62L hiCD25-CD44lo were obtained by sorting (BD FACS Aria). Additionally, for the RNAseq experiments, CD4 and CD8 naïve cells were isolated by sorting T cells from the Foxp3- IRES-GFP mice: CD4+CD62LhiCD25–CD44lo GFP(FOXP3)– and CD8+CD62LhiCD25– CD44lo GFP(FOXP3)– (antibodies were from Biolegend). In some cases, naïve CD4 cells were cultured in vitro under Th1 or Th2 polarizing conditions (3, 4). -

H4K16 Acetylation Marks Active Genes and Enhancers of Embryonic Stem Cells, but Does Not Alter Chromatin Compaction
Downloaded from genome.cshlp.org on October 5, 2021 - Published by Cold Spring Harbor Laboratory Press H4K16 acetylation marks active genes and enhancers of embryonic stem cells, but does not alter chromatin compaction Gillian Taylor1, Ragnhild Eskeland2, Betül Hekimoglu-Balkan1, Madapura M. Pradeepa1* and Wendy A Bickmore1* 1 MRC Human Genetics Unit, MRC Institute of Genetics and Molecular Medicine at University of Edinburgh, Crewe Road, Edinburgh EH4 2XU, UK 2Current address: Department of Molecular Biosciences, University of Oslo, N-0316 Oslo, Norway *Correspondence to: W. Bickmore or M.M. Pradeepa, MRC Human Genetics Unit, MRC IGMM, Crewe Road, Edinburgh EH4 2XU, UK Tel: +44 131 332 2471 Fax: +44 131 467 8456 Email:[email protected] or [email protected] Running head: H4K16 acetylation and long-range genome regulation Keywords: Chromatin compaction, embryonic stem cells, fluorescence in situ hybridization, histone acetylation, long-range regulation, 1 Downloaded from genome.cshlp.org on October 5, 2021 - Published by Cold Spring Harbor Laboratory Press Abstract Compared with histone H3, acetylation of H4 tails has not been well studied, especially in mammalian cells. Yet, H4K16 acetylation is of particular interest because of its ability to decompact nucleosomes in vitro and its involvement in dosage compensation in flies. Here we show that, surprisingly, loss of H4K16 acetylation does not alter higher-order chromatin compaction in vivo in mouse embryonic stem cells (ESCs). As well as peaks of acetylated H4K16 and Kat8/MOF histone acetyltransferase at the transcription start sites of expressed genes, we report that acetylation of H4K16 is a new marker of active enhancers in ESCs and that some enhancers are marked by H3K4me1, Kat8 and H4K16ac but not by acetylated H3K27 or p300/EP300, suggesting that they are novel EP300 independent regulatory elements. -

Histone Acetyltransferase Activity of MOF Is Required for MLL-AF9 Leukemogenesis Daria G
Published OnlineFirst February 15, 2017; DOI: 10.1158/0008-5472.CAN-16-2374 Cancer Tumor and Stem Cell Biology Research Histone Acetyltransferase Activity of MOF Is Required for MLL-AF9 Leukemogenesis Daria G. Valerio1,2, Haiming Xu1,2,3, Chun-Wei Chen1,2,3, Takayuki Hoshii1,2,3, Meghan E. Eisold1,2, Christopher Delaney1,2,3, Monica Cusan1,2, Aniruddha J. Deshpande1,4, Chun-Hao Huang2, Amaia Lujambio5, YuJun George Zheng6, Johannes Zuber7, Tej K. Pandita8, Scott W. Lowe2, and Scott A. Armstrong1,2,3 Abstract Chromatin-based mechanisms offer therapeutic targets in tumor cell genome. Rescue experiments with catalytically inac- acute myeloid leukemia (AML) that are of great current interest. tive mutants of MOF showed that its enzymatic activity was In this study, we conducted an RNAi-based screen to identify required to maintain cancer pathogenicity. In support of the druggable chromatin regulator–based targets in leukemias role of MOF in sustaining H4K16 acetylation, a small-molecule marked by oncogenic rearrangements of the MLL gene. In this inhibitor of the HAT component MYST blocked the growth of manner, we discovered the H4K16 histone acetyltransferase both murine and human MLL-AF9 leukemia cell lines. Further- (HAT) MOF to be important for leukemia cell growth. Condi- more, Mof inactivation suppressed leukemia development in tional deletion of Mof in a mouse model of MLL-AF9–driven an NUP98-HOXA9–driven AML model. Taken together, our leukemogenesis reduced tumor burden and prolonged host results establish that the HAT activity of MOF is required survival. RNA sequencing showed an expected downregulation to sustain MLL-AF9 leukemia and may be important for multiple of genes within DNA damage repair pathways that are con- AML subtypes. -
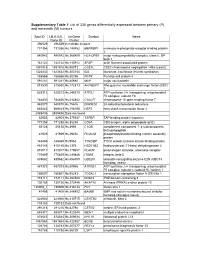
Supplementary Table 1 List of 335 Genes Differentially Expressed Between Primary (P) and Metastatic (M) Tumours
Supplementary Table 1 List of 335 genes differentially expressed between primary (P) and metastatic (M) tumours Spot ID I.M.A.G.E. UniGene Symbol Name Clone ID Cluster 296529 296529 In multiple clusters 731356 731356 Hs.140452 M6PRBP1 mannose-6-phosphate receptor binding protein 1 840942 840942 Hs.368409 HLA-DPB1 major histocompatibility complex, class II, DP beta 1 142122 142122 Hs.115912 AFAP actin filament associated protein 1891918 1891918 Hs.90073 CSE1L CSE1 chromosome segregation 1-like (yeast) 1323432 1323432 Hs.303154 IDS iduronate 2-sulfatase (Hunter syndrome) 788566 788566 Hs.80296 PCP4 Purkinje cell protein 4 591281 591281 Hs.80680 MVP major vault protein 815530 815530 Hs.172813 ARHGEF7 Rho guanine nucleotide exchange factor (GEF) 7 825312 825312 Hs.246310 ATP5J ATP synthase, H+ transporting, mitochondrial F0 complex, subunit F6 784830 784830 Hs.412842 C10orf7 chromosome 10 open reading frame 7 840878 840878 Hs.75616 DHCR24 24-dehydrocholesterol reductase 669443 669443 Hs.158195 HSF2 heat shock transcription factor 2 2485436 2485436 Data not found 82903 82903 Hs.370937 TAPBP TAP binding protein (tapasin) 771258 771258 Hs.85258 CD8A CD8 antigen, alpha polypeptide (p32) 85128 85128 Hs.8986 C1QB complement component 1, q subcomponent, beta polypeptide 41929 41929 Hs.39252 PICALM phosphatidylinositol binding clathrin assembly protein 148469 148469 Hs.9963 TYROBP TYRO protein tyrosine kinase binding protein 415145 415145 Hs.1376 HSD11B2 hydroxysteroid (11-beta) dehydrogenase 2 810017 810017 Hs.179657 PLAUR plasminogen activator, -

Variation in Protein Coding Genes Identifies Information
bioRxiv preprint doi: https://doi.org/10.1101/679456; this version posted June 21, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Animal complexity and information flow 1 1 2 3 4 5 Variation in protein coding genes identifies information flow as a contributor to 6 animal complexity 7 8 Jack Dean, Daniela Lopes Cardoso and Colin Sharpe* 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 Institute of Biological and Biomedical Sciences 25 School of Biological Science 26 University of Portsmouth, 27 Portsmouth, UK 28 PO16 7YH 29 30 * Author for correspondence 31 [email protected] 32 33 Orcid numbers: 34 DLC: 0000-0003-2683-1745 35 CS: 0000-0002-5022-0840 36 37 38 39 40 41 42 43 44 45 46 47 48 49 Abstract bioRxiv preprint doi: https://doi.org/10.1101/679456; this version posted June 21, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Animal complexity and information flow 2 1 Across the metazoans there is a trend towards greater organismal complexity. How 2 complexity is generated, however, is uncertain. Since C.elegans and humans have 3 approximately the same number of genes, the explanation will depend on how genes are 4 used, rather than their absolute number. -

Supplementary Materials
Supplementary materials Supplementary Table S1: MGNC compound library Ingredien Molecule Caco- Mol ID MW AlogP OB (%) BBB DL FASA- HL t Name Name 2 shengdi MOL012254 campesterol 400.8 7.63 37.58 1.34 0.98 0.7 0.21 20.2 shengdi MOL000519 coniferin 314.4 3.16 31.11 0.42 -0.2 0.3 0.27 74.6 beta- shengdi MOL000359 414.8 8.08 36.91 1.32 0.99 0.8 0.23 20.2 sitosterol pachymic shengdi MOL000289 528.9 6.54 33.63 0.1 -0.6 0.8 0 9.27 acid Poricoic acid shengdi MOL000291 484.7 5.64 30.52 -0.08 -0.9 0.8 0 8.67 B Chrysanthem shengdi MOL004492 585 8.24 38.72 0.51 -1 0.6 0.3 17.5 axanthin 20- shengdi MOL011455 Hexadecano 418.6 1.91 32.7 -0.24 -0.4 0.7 0.29 104 ylingenol huanglian MOL001454 berberine 336.4 3.45 36.86 1.24 0.57 0.8 0.19 6.57 huanglian MOL013352 Obacunone 454.6 2.68 43.29 0.01 -0.4 0.8 0.31 -13 huanglian MOL002894 berberrubine 322.4 3.2 35.74 1.07 0.17 0.7 0.24 6.46 huanglian MOL002897 epiberberine 336.4 3.45 43.09 1.17 0.4 0.8 0.19 6.1 huanglian MOL002903 (R)-Canadine 339.4 3.4 55.37 1.04 0.57 0.8 0.2 6.41 huanglian MOL002904 Berlambine 351.4 2.49 36.68 0.97 0.17 0.8 0.28 7.33 Corchorosid huanglian MOL002907 404.6 1.34 105 -0.91 -1.3 0.8 0.29 6.68 e A_qt Magnogrand huanglian MOL000622 266.4 1.18 63.71 0.02 -0.2 0.2 0.3 3.17 iolide huanglian MOL000762 Palmidin A 510.5 4.52 35.36 -0.38 -1.5 0.7 0.39 33.2 huanglian MOL000785 palmatine 352.4 3.65 64.6 1.33 0.37 0.7 0.13 2.25 huanglian MOL000098 quercetin 302.3 1.5 46.43 0.05 -0.8 0.3 0.38 14.4 huanglian MOL001458 coptisine 320.3 3.25 30.67 1.21 0.32 0.9 0.26 9.33 huanglian MOL002668 Worenine -

Appendix 2. Significantly Differentially Regulated Genes in Term Compared with Second Trimester Amniotic Fluid Supernatant
Appendix 2. Significantly Differentially Regulated Genes in Term Compared With Second Trimester Amniotic Fluid Supernatant Fold Change in term vs second trimester Amniotic Affymetrix Duplicate Fluid Probe ID probes Symbol Entrez Gene Name 1019.9 217059_at D MUC7 mucin 7, secreted 424.5 211735_x_at D SFTPC surfactant protein C 416.2 206835_at STATH statherin 363.4 214387_x_at D SFTPC surfactant protein C 295.5 205982_x_at D SFTPC surfactant protein C 288.7 1553454_at RPTN repetin solute carrier family 34 (sodium 251.3 204124_at SLC34A2 phosphate), member 2 238.9 206786_at HTN3 histatin 3 161.5 220191_at GKN1 gastrokine 1 152.7 223678_s_at D SFTPA2 surfactant protein A2 130.9 207430_s_at D MSMB microseminoprotein, beta- 99.0 214199_at SFTPD surfactant protein D major histocompatibility complex, class II, 96.5 210982_s_at D HLA-DRA DR alpha 96.5 221133_s_at D CLDN18 claudin 18 94.4 238222_at GKN2 gastrokine 2 93.7 1557961_s_at D LOC100127983 uncharacterized LOC100127983 93.1 229584_at LRRK2 leucine-rich repeat kinase 2 HOXD cluster antisense RNA 1 (non- 88.6 242042_s_at D HOXD-AS1 protein coding) 86.0 205569_at LAMP3 lysosomal-associated membrane protein 3 85.4 232698_at BPIFB2 BPI fold containing family B, member 2 84.4 205979_at SCGB2A1 secretoglobin, family 2A, member 1 84.3 230469_at RTKN2 rhotekin 2 82.2 204130_at HSD11B2 hydroxysteroid (11-beta) dehydrogenase 2 81.9 222242_s_at KLK5 kallikrein-related peptidase 5 77.0 237281_at AKAP14 A kinase (PRKA) anchor protein 14 76.7 1553602_at MUCL1 mucin-like 1 76.3 216359_at D MUC7 mucin 7,