Functional Genomic Screening Reveals Asparagine Dependence As A
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

TRYPTOPHANYL TRANSF'er RNA SYNTHETASE and EXPRESSION of the TRYPTOPHAN OPERON in the Trp8 MUTANTS of ESCHERICHIA COLI I N Additi
TRYPTOPHANYL TRANSF'ER RNA SYNTHETASE AND EXPRESSION OF THE TRYPTOPHAN OPERON IN THE trp8 MUTANTS OF ESCHERICHIA COLI KOREAKI ITO, SOTA HIRAGA AND TAKASHI WRA Institute for Virus Research, Kyoto Uniuersity, Kyoto, Japan Received November 15, 1968 INaddition to their role in protein synthesis, aminoacyl tRNA synthetases in bacteria have been shown to have a regulatory role in the formation of enzymes involved in the synthesis of corresponding amino acids. This has been demon- strated at least in the case of isoleucine and valine (EIDLICand NEIDHARDT1965; YANIV,JACOB and GROS1965) and histidine (SCHLESINGERand MAGASANIK1964; ROTH, ANTONand HARTMAN1966; ROTH and AMES1966). Transfer RNA (tRNA) also appears to be involved in the regulation of these systems (FREUND- LICH 1967; SILBERT,FINK and AMES1966). The expression of the genes determining the tryptophan biosynthetic enzymes, the tryptophan operon in Escherichia coli, is subject to repression by excess L-tryptophan, the end product of the pathway. The mutants of a regulatory gene, trpR, located outside of this operon, exhibit constitutive synthesis of the biosyn- thetic enzymes (COHENand JACOB1959). In addition, we have previously reported tryptophan auxotrophic mutants in which the known structural genes of the tryptophan operon remain intact. One of these mutants, designated trpiS5, has been mapped near strA on the chromosome (HIRAGA,ITO, HAMADA and YURA 1967a). In the present paper, we report on further characterization of the trpS5 mutant and discuss the possible role of the trpS gene in the regulation of the tryptophan operon. The results suggest that trpS, located between strA and maZA, is the structural gene for tryptophanyl tRNA synthetase, and that tryptophanyl tRNA synthetase is somehow involved in the repression of the tryptophan operon. -

Amino Acid Recognition by Aminoacyl-Trna Synthetases
www.nature.com/scientificreports OPEN The structural basis of the genetic code: amino acid recognition by aminoacyl‑tRNA synthetases Florian Kaiser1,2,4*, Sarah Krautwurst3,4, Sebastian Salentin1, V. Joachim Haupt1,2, Christoph Leberecht3, Sebastian Bittrich3, Dirk Labudde3 & Michael Schroeder1 Storage and directed transfer of information is the key requirement for the development of life. Yet any information stored on our genes is useless without its correct interpretation. The genetic code defnes the rule set to decode this information. Aminoacyl-tRNA synthetases are at the heart of this process. We extensively characterize how these enzymes distinguish all natural amino acids based on the computational analysis of crystallographic structure data. The results of this meta-analysis show that the correct read-out of genetic information is a delicate interplay between the composition of the binding site, non-covalent interactions, error correction mechanisms, and steric efects. One of the most profound open questions in biology is how the genetic code was established. While proteins are encoded by nucleic acid blueprints, decoding this information in turn requires proteins. Te emergence of this self-referencing system poses a chicken-or-egg dilemma and its origin is still heavily debated 1,2. Aminoacyl-tRNA synthetases (aaRSs) implement the correct assignment of amino acids to their codons and are thus inherently connected to the emergence of genetic coding. Tese enzymes link tRNA molecules with their amino acid cargo and are consequently vital for protein biosynthesis. Beside the correct recognition of tRNA features3, highly specifc non-covalent interactions in the binding sites of aaRSs are required to correctly detect the designated amino acid4–7 and to prevent errors in biosynthesis5,8. -

Amino Acid Chemistry
Handout 4 Amino Acid and Protein Chemistry ANSC 619 PHYSIOLOGICAL CHEMISTRY OF LIVESTOCK SPECIES Amino Acid Chemistry I. Chemistry of amino acids A. General amino acid structure + HN3- 1. All amino acids are carboxylic acids, i.e., they have a –COOH group at the #1 carbon. 2. All amino acids contain an amino group at the #2 carbon (may amino acids have a second amino group). 3. All amino acids are zwitterions – they contain both positive and negative charges at physiological pH. II. Essential and nonessential amino acids A. Nonessential amino acids: can make the carbon skeleton 1. From glycolysis. 2. From the TCA cycle. B. Nonessential if it can be made from an essential amino acid. 1. Amino acid "sparing". 2. May still be essential under some conditions. C. Essential amino acids 1. Branched chain amino acids (isoleucine, leucine and valine) 2. Lysine 3. Methionine 4. Phenyalanine 5. Threonine 6. Tryptophan 1 Handout 4 Amino Acid and Protein Chemistry D. Essential during rapid growth or for optimal health 1. Arginine 2. Histidine E. Nonessential amino acids 1. Alanine (from pyruvate) 2. Aspartate, asparagine (from oxaloacetate) 3. Cysteine (from serine and methionine) 4. Glutamate, glutamine (from α-ketoglutarate) 5. Glycine (from serine) 6. Proline (from glutamate) 7. Serine (from 3-phosphoglycerate) 8. Tyrosine (from phenylalanine) E. Nonessential and not required for protein synthesis 1. Hydroxyproline (made postranslationally from proline) 2. Hydroxylysine (made postranslationally from lysine) III. Acidic, basic, polar, and hydrophobic amino acids A. Acidic amino acids: amino acids that can donate a hydrogen ion (proton) and thereby decrease pH in an aqueous solution 1. -

S42003-019-0587-Z.Pdf
Corrected: Author Correction ARTICLE https://doi.org/10.1038/s42003-019-0587-z OPEN High-resolution crystal structure of human asparagine synthetase enables analysis of inhibitor binding and selectivity Wen Zhu 1,10, Ashish Radadiya 1, Claudine Bisson2,10, Sabine Wenzel 3, Brian E. Nordin4,11, 1234567890():,; Francisco Martínez-Márquez3, Tsuyoshi Imasaki 3,5, Svetlana E. Sedelnikova2, Adriana Coricello 1,6,7, Patrick Baumann 1, Alexandria H. Berry8, Tyzoon K. Nomanbhoy4, John W. Kozarich 4, Yi Jin 1, David W. Rice 2, Yuichiro Takagi 3 & Nigel G.J. Richards 1,9 Expression of human asparagine synthetase (ASNS) promotes metastatic progression and tumor cell invasiveness in colorectal and breast cancer, presumably by altering cellular levels of L-asparagine. Human ASNS is therefore emerging as a bona fide drug target for cancer therapy. Here we show that a slow-onset, tight binding inhibitor, which exhibits nanomolar affinity for human ASNS in vitro, exhibits excellent selectivity at 10 μM concentration in HCT- 116 cell lysates with almost no off-target binding. The high-resolution (1.85 Å) crystal structure of human ASNS has enabled us to identify a cluster of negatively charged side chains in the synthetase domain that plays a key role in inhibitor binding. Comparing this structure with those of evolutionarily related AMP-forming enzymes provides insights into intermolecular interactions that give rise to the observed binding selectivity. Our findings demonstrate the feasibility of developing second generation human ASNS inhibitors as lead compounds for the discovery of drugs against metastasis. 1 School of Chemistry, Cardiff University, Cardiff, UK. 2 Department of Molecular Biology and Biotechnology, University of Sheffield, Sheffield, UK. -

Stimulation Effects of Foliar Applied Glycine and Glutamine Amino Acids
Open Agriculture. 2019; 4: 164–172 Research Article Yaghoub Aghaye Noroozlo, Mohammad Kazem Souri*, Mojtaba Delshad Stimulation Effects of Foliar Applied Glycine and Glutamine Amino Acids on Lettuce Growth https://doi.org/10.1515/opag-2019-0016 received June 27, 2018; accepted January 20, 2019 1 Introduction Abstract: Amino acids have various roles in plant In biology, amino acids have vital roles in cell life. Amino metabolism, and exogenous application of amino acids acids are among the most important primary metabolites may have benefits and stimulation effects on plant growth within the plant cells. However, they are frequently and quality. In this study, the growth and nutrient uptake regarded as secondary metabolites, particularly in the of Romain lettuce (Lactuca sativa subvar Sahara) were case of proline, glycine and betaine amino acids. Many evaluated under spray of glycine or glutamine at different physiochemical characteristics of plant cells, tissues and concentrations of 0 (as control), 250, 500 and 1000 organs are influenced by the presence of amino acids (Rai mg.L-1, as well as a treatment of 250 mg.L-1 glycine+250 2002; Marschner 2011). They are the building units of mg.L-1 glutamine. The results showed that there was proteins, as the main component of living cells that have significant increase in leaf total chlorophyll content under vital roles in many cell metabolic reactions (Kielland 1994; Gly250+Glu250, Gly250 and Glu1000 mg.L-1treatments, and Rainbird et al. 1984; Jones and Darrah 1993). In addition, in leaf carotenoids content under 250 mg.L-1 glutamine amino acids have various important biological functions spray compared with the control plants. -

Novel Derivatives of Nicotinamide Adenine Dinucleotide (NAD) and Their Biological Evaluation Against NAD- Consuming Enzymes
Novel derivatives of nicotinamide adenine dinucleotide (NAD) and their biological evaluation against NAD- Consuming Enzymes Giulia Pergolizzi University of East Anglia School of Pharmacy Thesis submitted for the degree of Doctor of Philosophy July, 2012 © This copy of the thesis has been supplied on condition that anyone who consults it is understood to recognise that its copyright rests with the author and that use of any information derived there from must be in accordance with current UK Copyright Law. In addition, any quotation or extract must include full attribution. ABSTRACT Nicotinamide adenine dinucleotide (β-NAD+) is a primary metabolite involved in fundamental biological processes. Its molecular structure with characteristic functional groups, such as the quaternary nitrogen of the nicotinamide ring, and the two high- energy pyrophosphate and nicotinamide N-glycosidic bonds, allows it to undergo different reactions depending on the reactive moiety. Well known as a redox substrate owing to the redox properties of the nicotinamide ring, β-NAD+ is also fundamental as a substrate of NAD+-consuming enzymes that cleave either high-energy bonds to catalyse their reactions. In this study, a panel of novel adenine-modified NAD+ derivatives was synthesized and biologically evaluated against different NAD+-consuming enzymes. The synthesis of NAD+ derivatives, modified in position 2, 6 or 8 of the adenine ring with aryl/heteroaryl groups, was accomplished by Suzuki-Miyaura cross-couplings. Their biological activity as inhibitors and/or non-natural substrates was assessed against a selected range of NAD+-consuming enzymes. The fluorescence of 8-aryl/heteroaryl NAD+ derivatives allowed their use as biochemical probes for the development of continuous biochemical assays to monitor NAD+-consuming enzyme activities. -

Nucleotide Base Coding and Am1ino Acid Replacemients in Proteins* by Emil L
VOL. 48, 1962 BIOCHEMISTRY: E. L. SAIITH 677 18 Britten, R. J., and R. B. Roberts, Science, 131, 32 (1960). '9 Crestfield, A. M., K. C. Smith, and F. WV. Allen, J. Biol. Chem., 216, 185 (1955). 20 Gamow, G., Nature, 173, 318 (1954). 21 Brenner, S., these PROCEEDINGS, 43, 687 (1957). 22 Nirenberg, M. WV., J. H. Matthaei, and 0. WV. Jones, unpublished data. 23 Crick, F. H. C., L. Barnett, S. Brenner, and R. J. Watts-Tobin, Nature, 192, 1227 (1961). 24 Levene, P. A., and R. S. Tipson, J. Biol. Ch-nn., 111, 313 (1935). 25 Gierer, A., and K. W. Mundry, Nature, 182, 1437 (1958). 2' Tsugita, A., and H. Fraenkel-Conrat, J. Mllot. Biol., in press. 27 Tsugita, A., and H. Fraenkel-Conrat, personal communication. 28 Wittmann, H. G., Naturwissenschaften, 48, 729 (1961). 29 Freese, E., in Structure and Function of Genetic Elements, Brookhaven Symposia in Biology, no. 12 (1959), p. 63. NUCLEOTIDE BASE CODING AND AM1INO ACID REPLACEMIENTS IN PROTEINS* BY EMIL L. SMITHt LABORATORY FOR STUDY OF HEREDITARY AND METABOLIC DISORDERS AND THE DEPARTMENTS OF BIOLOGICAL CHEMISTRY AND MEDICINE, UNIVERSITY OF UTAH COLLEGE OF MEDICINE Communicated by Severo Ochoa, February 14, 1962 The problem of which bases of messenger or template RNA' specify the coding of amino acids in proteins has been largely elucidated by the use of synthetic polyri- bonucleotides.2-7 For these triplet nucleotide compositions (Table 1), it is of in- terest to examine some of the presently known cases of amino acid substitutions in polypeptides or proteins of known structure. -

Feedback Inhibition of Fully Unadenylylated Glutamine
Proc. Natl. Acad. Sci. USA Vol. 90, pp. 4996-5000, June 1993 Biochemistry Feedback inhibition of fully unadenylylated glutamine synthetase from Salmonella typhimurium by glycine, alanine, and serine (x-ray crystaflography/protein structure/effector binding) SHWU-HUEY LIAW, CLARK PAN, AND DAVID EISENBERG* Molecular Biology Institute and Department of Chemistry and Biochemistry, University of California, Los Angeles, CA 90024 Contributed by David Eisenberg, December 28, 1992 ABSTRACT Bacterial glutamine synthetase (GS; EC the binding of L-glutamate to GS was measured in the 6.3.1.2) was previously shown to be inhibited by nine end presence of ADP and Pi (6, 9). Citing these studies and products of glutamine metabolism. Here we present four unpublished data, Stadtman and Ginsburg (2) concluded crystal structures ofGS, complexed with the substrate Glu and "there are separate sites on the enzyme for alanine, tryp- with each of three feedback inhibitors. The GS of the present tophan, histidine, AMP, and CTP, whereas mutually exclu- study is from Salmonela typhimurium, with Mn2+ ions bound, sive binding occurs between glycine, serine, and alanine" and is fully unadenylylated. From Fourier difference maps, we (P. Z. Smymiotis and E. R. Stadtman, unpublished data find that L-serine, L-alanine, and glycine bind at the site of the cited in review reference 2). And Rhee et al. (11) reviewed substrate L-glutamate. In our model, these four amino acids further evidence for separate sites of inhibition. In short, bind with the atoms they share in common (the "main chain" feedback inhibition ofGS is a complicated regulatory system, +NH3-CH-COO-) in the same positions. -

An Integrated Meta-Analysis of Peripheral Blood Metabolites and Biological Functions in Major Depressive Disorder
Molecular Psychiatry https://doi.org/10.1038/s41380-020-0645-4 ARTICLE An integrated meta-analysis of peripheral blood metabolites and biological functions in major depressive disorder 1,2,3 1,2,3 1,2,3 1,3 1,3 4,5 1,3 1,3 Juncai Pu ● Yiyun Liu ● Hanping Zhang ● Lu Tian ● Siwen Gui ● Yue Yu ● Xiang Chen ● Yue Chen ● 1,2,3 1,3 1,3 1,3 1,3 1,2,3 Lining Yang ● Yanqin Ran ● Xiaogang Zhong ● Shaohua Xu ● Xuemian Song ● Lanxiang Liu ● 1,2,3 1,3 1,2,3 Peng Zheng ● Haiyang Wang ● Peng Xie Received: 3 June 2019 / Revised: 24 December 2019 / Accepted: 10 January 2020 © The Author(s) 2020. This article is published with open access Abstract Major depressive disorder (MDD) is a serious mental illness, characterized by high morbidity, which has increased in recent decades. However, the molecular mechanisms underlying MDD remain unclear. Previous studies have identified altered metabolic profiles in peripheral tissues associated with MDD. Using curated metabolic characterization data from a large sample of MDD patients, we meta-analyzed the results of metabolites in peripheral blood. Pathway and network analyses were then performed to elucidate the biological themes within these altered metabolites. We identified 23 differentially 1234567890();,: 1234567890();,: expressed metabolites between MDD patients and controls from 46 studies. MDD patients were characterized by higher levels of asymmetric dimethylarginine, tyramine, 2-hydroxybutyric acid, phosphatidylcholine (32:1), and taurochenode- soxycholic acid and lower levels of L-acetylcarnitine, creatinine, L-asparagine, L-glutamine, linoleic acid, pyruvic acid, palmitoleic acid, L-serine, oleic acid, myo-inositol, dodecanoic acid, L-methionine, hypoxanthine, palmitic acid, L-tryptophan, kynurenic acid, taurine, and 25-hydroxyvitamin D compared with controls. -

Single-Site Glycine-Specific Labeling of Proteins
ARTICLE https://doi.org/10.1038/s41467-019-10503-7 OPEN Single-site glycine-specific labeling of proteins Landa Purushottam1, Srinivasa Rao Adusumalli1, Usha Singh2, V.B. Unnikrishnan1, Dattatraya Gautam Rawale1, Mansi Gujrati2, Ram Kumar Mishra2 & Vishal Rai 1 Labeling of native proteins invites interest from diverse segments of science. However, there remains the significant unmet challenge in precise labeling at a single site of a protein. Here, we report the site-specific labeling of natural or easy-to-engineer N-terminus Gly in proteins with remarkable efficiency and selectivity. The method generates a latent nucleophile from 1234567890():,; N-terminus imine that reacts with an aldehyde to deliver an aminoalcohol under physiological conditions. It differentiates N-Gly as a unique target amongst other proteinogenic amino acids. The method allows single-site labeling of proteins in isolated form and extends to lysed cells. It administers an orthogonal aldehyde group primed for late-stage tagging with an affinity tag, 19F NMR probe, and a fluorophore. A user-friendly protocol delivers analytically pure tagged proteins. The mild reaction conditions do not alter the structure and function of the protein. The cellular uptake of fluorophore-tagged insulin and its ability to activate the insulin-receptor mediated signaling remains unperturbed. 1 Department of Chemistry, Indian Institute of Science Education and Research (IISER) Bhopal, Bhopal 462066, India. 2 Department of Biological Sciences, Indian Institute of Science Education and Research -

Effects of Glycine on Collagen, PDGF, and EGF Expression in Model of Oral Mucositis
nutrients Article Effects of Glycine on Collagen, PDGF, and EGF Expression in Model of Oral Mucositis Odara Maria de Sousa Sá 1,* , Nilza Nelly Fontana Lopes 2, Maria Teresa Seixas Alves 3 and Eliana Maria Monteiro Caran 4 1 Department of Pediatrics, Federal University of São Paulo, São Paulo 04023-062, Brazil 2 Former Head Division of Dentistry, Pediatric Oncology Institute, São Paulo 04023-062, Brazil; nnfl[email protected] 3 Department of Pathology, Federal University of São Paulo, São Paulo 04023-062, Brazil; [email protected] 4 Department of Pediatrics, IOP/GRAACC Medical School of Federal University of São Paulo, São Paulo 04023-062, Brazil; [email protected] * Correspondence: [email protected]; Tel.: +55-086-99932-5493 Received: 10 July 2018; Accepted: 5 September 2018; Published: 12 October 2018 Abstract: Oral mucositis is frequently a toxic effect of chemotherapeutic and/or radiotherapeutic treatment, resulting from complex multifaceted biological events involving DNA damage. The clinical manifestations have a negative impact on the life quality of cancer patients. Preventive measures and curative treatment of mucositis are still not well established. The glycine has anti-inflammatory, immunomodulatory, and cytoprotective actions, being a potential therapeutic in mucositis. The objective was to evaluate the effects of glycine on the expression of collagen and growth factors, platelet and epidermal in a hamster model oral mucositis. The mucositis was induced by the protocol of Sonis. There were 40 hamsters used, divided into two groups: Group I-control; Group II-supplemented with 5% intraperitoneal glycine, 2.0 mg/g diluted in hepes. Histopathological sections were used to perform the immune-histochemical method, the evaluation of collagen expression, and the growth factors: Epidermal growth factor (EGF) and platelet (PDGF). -
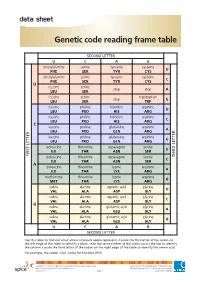
Genetic Code Reading Frame Table
data sheet Genetic code reading frame table SECOND LETTER U C A G phenylalanine serine tyrosine cysteine U PHE SER TYR CYS phenylalanine serine tyrosine cysteine C PHE SER TYR CYS U leucine serine stop stop A LEU SER leucine serine tryptophan stop G LEU SER TRP leucine proline histidine arginine U LEU PRO HIS ARG leucine proline histidine arginine C LEU PRO HIS ARG C leucine proline glutamine arginine A LEU PRO GLN ARG leucine proline glutamine arginine G LEU PRO GLN ARG isoleucine threonine asparagine serine U ILE THR ASN SER FIRST LETTER isoleucine threonine asparagine serine LETTERTHIRD C ILE THR ASN SER A isoleucine threonine lysine arginine A ILE THR LYS ARG methionine threonine lysine arginine G MET THR LYS ARG valine alanine aspartic acid glycine U VAL ALA ASP GLY valine alanine aspartic acid glycine C VAL ALA ASP GLY G valine alanine glutamic acid glycine A VAL ALA GLU GLY valine alanine glutamic acid glycine G VAL ALA GLU GLY U C A G SECOND LETTER Use this table to find out what amino acid each codon represents. Locate the first letter of the codon on the left edge of the table to identify a block, then the second letter of the codon across the top to identify the column. Locate the third letter of the codon on the right edge of the table to identify the amino acid. For example, the codon ‘CAU’ codes for histidine (HIS). ast0812 | Proteins 3: Genetic code reading frame table (data sheet) developed for the Department of Education WA © The University of Western Australia 2012 for conditions of use see spice.wa.edu.au/usage version 1.0 page 1 Licensed for NEALS.