Chemical Contaminants 201 Continuing Education Professional Development Course
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Standard X-Ray Diffraction Powder Patterns
NBS MONOGRAPH 25 — SECTION 1 Standard X-ray Diffraction U.S. DEPARTMENT OF COMMERCE NATIONAL BUREAU OF STANDARDS THE NATIONAL BUREAU OF STANDARDS Functions and Activities The functions of the National Bureau of Standards are set forth in the Act of Congress, March 3, 1901, as amended by Congress in Public Law 619, 1950. These include the development and maintenance of the national standards of measurement and the provision of means and methods for making measurements consistent with these standards; the determination of physical constants and properties of materials; the development of methods and instruments for testing materials, devices, and structures; advisory services to government agencies on scien- tific and technical problems; invention and development of devices to serve special needs of the Government; and the development of standard practices, codes, and specifications. The work includes basic and applied research, development, engineering, instrumentation, testing, evaluation, calibration services, and various consultation and information services. Research projects are also performed for other government agencies when the work relates to and supplements the basic program of the Bureau or when the Bureau's unique competence is required. The scope of activities is suggested by the listing of divisions and sections on the inside of the back cover. Publications The results of the Bureau's research are published either in the Bureau's own series of publications or in the journals of professional and scientific societies. The Bureau itself publishes three periodicals available from the Government Printing Office: The Journal of Research, published in four separate sections, presents complete scientific and technical papers; the Technical News Bulletin presents summary and preliminary reports on work in progress; and Basic Radio Propagation Predictions provides data for determining the best frequencies to use for radio communications throughout the world. -

Amarsinha D. Nikam Vital Force Is Oxygen Extrait Du Livre Vital Force Is Oxygen De Amarsinha D
Amarsinha D. Nikam Vital Force is Oxygen Extrait du livre Vital Force is Oxygen de Amarsinha D. Nikam Éditeur : B. Jain http://www.editions-narayana.fr/b9138 Sur notre librairie en ligne vous trouverez un grand choix de livres d'homéopathie en français, anglais et allemand. Reproduction des extraits strictement interdite. Narayana Verlag GmbH, Blumenplatz 2, D-79400 Kandern, Allemagne Tel. +33 9 7044 6488 Email [email protected] http://www.editions-narayana.fr Oxygen Oxygen is derived from the Greek word, where oxys means acid, literally sharp from the taste of acids and genes means producer, literally begetter. It is the element with atomic number 8 and represented by the symbol 'O'. It is a highly reactive non-metallic period 2 element that readily forms compounds (notably oxides) with almost all other elements. Oxygen is the third most abundant element in the universe by mass after hydrogen and helium. Diatomic oxygen gas constitutes 21% of the volume of air. Water is the most familiar oxygen compound. OXYGEN HISTORY Oxygen makes up 21% of the atmosphere we breathe, but it was not discovered as a separate gas until the late i8th century. Oxygen was independently discovered by Carl Wilhelm Scheele, in Uppsala in 1773 or earlier and by Joseph Priestley in Wiltshire, in 1774. The name oxygen was coined in 1777 by Antoine Lavoisier. Although oxygen plays a life-supporting role, it took Narayana Verlag, 79400 Kandern Tel.: 0049 7626 974 970 0 Excerpt from Dr. Amarsinha D.Nikam: Vital Force is Oxygen about 150 years for the gas to be used in a proper manner for patients. -

Materials Design and Band Gap Engineering of Complex
University of Louisville ThinkIR: The University of Louisville's Institutional Repository Electronic Theses and Dissertations 8-2016 Materials design and band gap engineering of complex nanostructures using a semi-empirical approach : low dimensional boron nanostructures, h-BN sheet with graphene domains and holey graphene. Cherno Baba Kah Follow this and additional works at: https://ir.library.louisville.edu/etd Part of the Condensed Matter Physics Commons Recommended Citation Kah, Cherno Baba, "Materials design and band gap engineering of complex nanostructures using a semi- empirical approach : low dimensional boron nanostructures, h-BN sheet with graphene domains and holey graphene." (2016). Electronic Theses and Dissertations. Paper 2491. https://doi.org/10.18297/etd/2491 This Doctoral Dissertation is brought to you for free and open access by ThinkIR: The University of Louisville's Institutional Repository. It has been accepted for inclusion in Electronic Theses and Dissertations by an authorized administrator of ThinkIR: The University of Louisville's Institutional Repository. This title appears here courtesy of the author, who has retained all other copyrights. For more information, please contact [email protected]. MATERIALS DESIGN AND BAND GAP ENGINEERING OF COMPLEX : LOW NANOSTRUCTURES USING A SEMI-EMPIRICAL APPROACH , h DIMENSIONAL BORON NANOSTRUCTURES - BN SHEET WITH GRAPHENE DOMAINS AND HOLEY GRAPHENE By Cherno Baba Kah M.S, University of Louisville, 2013 M.S, Clark Atlanta University, 2011 B.S, University of The Gambia, -

Environment and Ecology BHM 403 Techno India
Environment and Ecology BHM 403 Techno India ENVIRONMENT AND ECOLOGY BHM 403 (2008 -2017) PREPARED BY ANIS CHATTOPADHYAY ASST. PROFESSOR TECHNO INDIA EM-4/1, SECTOR –V, SALT LAKE KOLKATA -700091 1 Environment and Ecology BHM 403 Techno India 2008 GROUP – A ( Multiple Choice Type Questions ) 1. Choose correct answer from the given alternatives in each of the following questions : 10x1 = 10 i) Environmental Studies involve studies of a) evolution of life b) all aspects of human environment c) nitrogen cycle d) oxygen cycle. b ii) "Itai itai' disease is caused by a) Zinc b) Cadmium c) Mercury d) Iron. b iii) El Nino starts from a) Mediterranean coast b) Chinese coast c) South American cost d) Indian cost. C iv) The Greenhouse effect is due to a) Carbon dioxide, water vapour, methane and chlorofluorocarbons b) Nitrogen oxide c) Sulphur oxide d) Carbon monoxide. A v) The Ganga pollution is due to dumping of a) domestic and industrial sewages b) waste from forest c) food waste d) hospital waste. A vi) Biotic factor of ecosystem is a) Solar energy b) Temperature c) Soil d) Plants and animals. D vii) Medha Patkar is involved in a) Chipko movement b) Silent Valley movement c) Narmada Bachao movement d) none of these. C viii) The "Kyoto Protocol" is related with a) air pollution b) noise pollution c) water pollution d) none of these. A ix) BOD stands as a) Biochemical Oxygen Demand b) Biological Oxygen Demand c) Biggest Oxygen Demand d) Blown Out Dose. B x) The protective shield for life on earth is a) Carbon dioxide b) Ozone c) Oxygen d) Hydrogen. -

The Chemistry of Carbene-Stabilized
THE CHEMISTRY OF CARBENE-STABILIZED MAIN GROUP DIATOMIC ALLOTROPES by MARIHAM ABRAHAM (Under the Direction of Gregory H. Robinson) ABSTRACT The syntheses and molecular structures of carbene-stabilized arsenic derivatives of 1 1 i 1 1 AsCl3 (L :AsCl3 (1); L : = :C{N(2,6- Pr2C6H3)CH}2), and As2 (L :As–As:L (2)), are presented herein. The potassium graphite reduction of 1 afforded the carbene-stabilized diarsenic complex, 2. Notably, compound 2 is the first Lewis base stabilized diatomic molecule of the Group 13–15 elements, in the formal oxidation state of zero, in the fourth period or lower of the Periodic Table. Compound 2 contains one As–As σ-bond and two lone pairs of electrons on each arsenic atom. In an effort to study the chemistry of the electron-rich compound 2, it was combined with an electron-deficient Lewis acid, GaCl3. The addition of two equivalents of GaCl3 to 2 resulted in one-electron oxidation of 2 to 1 1 •+ – •+ – give [L :As As:L ] [GaCl4] (6 [GaCl4] ). Conversely, the addition of four equivalents of GaCl3 to 2 resulted in two- electron oxidation of 2 to give 1 1 2+ – 2+ – •+ [L :As=As:L ] [GaCl4 ]2 (6 [GaCl4 ]2). Strikingly, 6 represents the first arsenic radical to be structurally characterized in the solid state. The research project also explored the reactivity of carbene-stabilized disilicon, (L1:Si=Si:L1 (7)), with borane. The reaction of 7 with BH3·THF afforded two unique compounds: one containing a parent silylene (:SiH2) unit (8), and another containing a three-membered silylene ring (9). -

Physical and Chemical Properties of Germanium
Physical And Chemical Properties Of Germanium Moneyed and amnesic Erasmus fertilise her fatuousness revitalise or burrow incommunicatively. Creditable Petr still climbs: regarding and lissome Lazarus bully-off quite punctiliously but slums her filoplume devotedly. Zane still defilade venomous while improvident Randell bloodiest that wonderers. Do you for this context of properties and physical explanation of Silicon is sincere to metals in its chemical behaviour. Arsenic is extremely toxic, RS, carbon is the tongue one considered a full nonmetal. In nature, which name a widely used azo dye. Basic physical and chemical properties of semiconductors are offset by the energy gap between valence conduction! Other metalloids on the periodic table are boron, Batis ZB, only Germanium and Antimony would be considered metals for the purposes of nomenclature. Storage temperature: no restrictions. At room temperature, the semiconducting elements are primarily nonmetallic in character. This application requires Javascript. It has also new found in stars and already the atmosphere of Jupiter. Wellings JS, it is used as an eyewash and insecticide. He has studied in Spain and Hungary and authored many research articles published in indexed journals and books. What are oral health benefits of pumpkins? The material on this site may not be reproduced, germanium, the radiation emitted from an active device makes it locatable. Classify each statement as an extensive property must an intensive property. In germanium and physical chemical properties of the border lines from the! The most electronegative elements are at the nod in the periodic table; these elements often react as oxidizing agents. Atomic Volume and Allotropy of the Elements. -

Removal of 2, 4-Dinitrophenol by Ferrate
University of Central Florida STARS Electronic Theses and Dissertations, 2004-2019 2008 Removal Of 2, 4-dinitrophenol By Ferrate Gianna Cooley University of Central Florida Part of the Environmental Engineering Commons Find similar works at: https://stars.library.ucf.edu/etd University of Central Florida Libraries http://library.ucf.edu This Masters Thesis (Open Access) is brought to you for free and open access by STARS. It has been accepted for inclusion in Electronic Theses and Dissertations, 2004-2019 by an authorized administrator of STARS. For more information, please contact [email protected]. STARS Citation Cooley, Gianna, "Removal Of 2, 4-dinitrophenol By Ferrate" (2008). Electronic Theses and Dissertations, 2004-2019. 3619. https://stars.library.ucf.edu/etd/3619 REMOVAL OF 2, 4-DINITROPHENOL BY FERRATE by GIANNA GRIFFITH COOLEY B.S. Loyola Univeristy New Orleans, 2002 A thesis submitted in partial fulfillment of the requirements for the degree of Master of Environmental Engineering in the Department of Civil and Environmental Engineering in the College of Engineering and Computer Science at the University of Central Florida Orlando, Florida Fall Term 2008 Major Professor: Debra R. Reinhart © 2008 Gianna Griffith Cooley ii ABSTRACT VI 2- Ferrate (molecular formula, Fe O4 ) has been studied increasingly since the 1970s as a disinfectant and coagulant for domestic wastewater and also as an oxidant for industrial wastewaters (Murmann and Roginson, 1974, Gilbert et al., 1978, Kazama, 1994, Jiang et al., 2002, and Sharmaet al., 2005). This research was performed to explore whether ferrate could possibly be used as chemical treatment for industrial wastewaters from plastic, chemical, dye, soap, and wood stain producing plants that contain 2, 4-Dinitrophenol (DNP). -

Lecture 25: Main Group Chemistry
Lecture 23: Main Group Chemistry An Introduction We have spent the better part of the school year developing physical models to describe atomic structure, chemical bonding and the thermodynamics and kinetics physical and chemical phenomena. In all of that, however, we have been just as content to use generic symbols like A + B Æ C + D or to describe a weak acid as HA, as to actually assign it a real chemical structure with real chemical reactivity. In this chapter we right this wrong, working systematically through the main groups of the periodic table, learning for each group and particularly significant elements in those groups: • The natural forms in which the elements are found, including the hydrides and oxides • The famous chemical reactions that involve the chemical compounds containing those elements • The important practical uses of the compounds containing those elements. Common themes in Main Group Chemistry: We must have learned something useful during the year that will help to explain the chemistry of the main group elements. Here are some important themes that appear to explain pretty much everything that happens 1. The number of valence electrons in atoms drive the chemistry of a compound. And the main group number corresponds to the number of valence electrons 2. Compounds formed by main group elements pretty much always satisfy the octet rule if they can, achieving noble gas- like electronic configurations 3. There are trends in the periodic table that are pretty much determined by the effective nuclear charge and overall number of electrons in an atom. These trends are: a) atomic radius decreases moving to the right and up the periodic table b) ionization energy increases moving to the right and up the periodic table c) electronegativity increases to the right and up the periodic table d) polarizability increases to the right and down the periodic table e) non-metallic character increases to the right and up the periodic table 4. -

Operation Permit Application
Un; iy^\ tea 0 9 o Operation Permit Application Located at: 2002 North Orient Road Tampa, Florida 33619 (813) 623-5302 o Training Program TRAINING PROGRAM for Universal Waste & Transit Orient Road Tampa, Florida m ^^^^ HAZARDOUS WAb 1 P.ER^AlTTlNG TRAINING PROGRAM MASTER INDEX CHAPTER 1: Introduction Tab A CHAPTER 2: General Safety Manual Tab B CHAPTER 3: Protective Clothing Guide Tab C CHAPTER 4: Respiratory Training Program Tab D APPENDIX 1: Respiratory Training Program II Tab E CHAPTER 5: Basic Emergency Training Guide Tab F CHAPTER 6: Facility Operations Manual Tab G CHAPTER 7: Land Ban Certificates Tab H CHAPTER 8: Employee Certification Statement Tab. I CHAPTER ONE INTRODUCTION prepared by Universal Waste & Transit Orient Road Tampa Florida Introducti on STORAGE/TREATMENT PERSONNEL TRAINING PROGRAM All personnel involved in any handling, transportation, storage or treatment of hazardous wastes are required to start the enclosed training program within one-week after the initiation of employment at Universal Waste & Transit. This training program includes the following: Safety Equipment Personnel Protective Equipment First Aid & CPR Waste Handling Procedures Release Prevention & Response Decontamination Procedures Facility Operations Facility Maintenance Transportation Requirements Recordkeeping We highly recommend that all personnel involved in the handling, transportation, storage or treatment of hazardous wastes actively pursue additional technical courses at either the University of South Florida, or Tampa Junior College. Recommended courses would include general chemistry; analytical chemistry; environmental chemistry; toxicology; and additional safety and health related topics. Universal Waste & Transit will pay all registration, tuition and book fees for any courses which are job related. The only requirement is the successful completion of that course. -

Standard Thermodynamic Properties of Chemical
STANDARD THERMODYNAMIC PROPERTIES OF CHEMICAL SUBSTANCES ∆ ° –1 ∆ ° –1 ° –1 –1 –1 –1 Molecular fH /kJ mol fG /kJ mol S /J mol K Cp/J mol K formula Name Crys. Liq. Gas Crys. Liq. Gas Crys. Liq. Gas Crys. Liq. Gas Ac Actinium 0.0 406.0 366.0 56.5 188.1 27.2 20.8 Ag Silver 0.0 284.9 246.0 42.6 173.0 25.4 20.8 AgBr Silver(I) bromide -100.4 -96.9 107.1 52.4 AgBrO3 Silver(I) bromate -10.5 71.3 151.9 AgCl Silver(I) chloride -127.0 -109.8 96.3 50.8 AgClO3 Silver(I) chlorate -30.3 64.5 142.0 AgClO4 Silver(I) perchlorate -31.1 AgF Silver(I) fluoride -204.6 AgF2 Silver(II) fluoride -360.0 AgI Silver(I) iodide -61.8 -66.2 115.5 56.8 AgIO3 Silver(I) iodate -171.1 -93.7 149.4 102.9 AgNO3 Silver(I) nitrate -124.4 -33.4 140.9 93.1 Ag2 Disilver 410.0 358.8 257.1 37.0 Ag2CrO4 Silver(I) chromate -731.7 -641.8 217.6 142.3 Ag2O Silver(I) oxide -31.1 -11.2 121.3 65.9 Ag2O2 Silver(II) oxide -24.3 27.6 117.0 88.0 Ag2O3 Silver(III) oxide 33.9 121.4 100.0 Ag2O4S Silver(I) sulfate -715.9 -618.4 200.4 131.4 Ag2S Silver(I) sulfide (argentite) -32.6 -40.7 144.0 76.5 Al Aluminum 0.0 330.0 289.4 28.3 164.6 24.4 21.4 AlB3H12 Aluminum borohydride -16.3 13.0 145.0 147.0 289.1 379.2 194.6 AlBr Aluminum monobromide -4.0 -42.0 239.5 35.6 AlBr3 Aluminum tribromide -527.2 -425.1 180.2 100.6 AlCl Aluminum monochloride -47.7 -74.1 228.1 35.0 AlCl2 Aluminum dichloride -331.0 AlCl3 Aluminum trichloride -704.2 -583.2 -628.8 109.3 91.1 AlF Aluminum monofluoride -258.2 -283.7 215.0 31.9 AlF3 Aluminum trifluoride -1510.4 -1204.6 -1431.1 -1188.2 66.5 277.1 75.1 62.6 AlF4Na Sodium tetrafluoroaluminate -

Physical Sciences Applications 09:00 - 12:00 Thursday, 26Th November, 2020 Track Physical Sciences Applications Presentation Type Oral Presentation
Physical Sciences Applications 09:00 - 12:00 Thursday, 26th November, 2020 Track Physical Sciences Applications Presentation type Oral Presentation 09:00 - 09:15 192 Quantifying ordering phenomena in lanthanum barium ferrate at the atomic scale Judith Lammer1, Christian Berger2, Stefan Löffler3, Daniel Knez1, Paolo Longo4, Edith Bucher2, Gerald Kothleitner1, Andreas Egger2, Rotraut Merkle5, Werner Sitte2, Joachim Maier5, Werner Grogger1 1Institute of Electron Microscopy and Nanoanalysis (FELMI), Graz University of Technology & Graz Centre for Electron Microscopy (ZFE), Graz, Austria. 2Chair of Physical Chemistry, Montanuniversitaet Leoben, Leoben, Austria. 3University Service Centre for Transmission Electron Microscopy (USTEM), TU Wien, Vienna, Austria. 4Gatan Inc., Pleasanton, USA. 5Max Planck Institute for Solid State Research, Stuttgart, Germany Abstract Text Within this abstract we present a high-resolution chemical analysis of the second order Ruddlesden-Popper phase Ba1.1La1.9Fe2O7 using EDX and EELS in STEM, as well as simulations for quantifying the lanthanum and barium concentration on each atom column. Lanthanum barium ferrates have auspicious properties as triple conducting oxides (proton-, oxygen ion- and electron-conducting) and therefore are promising new materials for future applications in protonic ceramic fuel cells, electrolyser cells or membranes for hydrogen separation [1]. However, mass and charge transport as well as defect chemistry are only partially understood at this point. These properties are influenced by the -
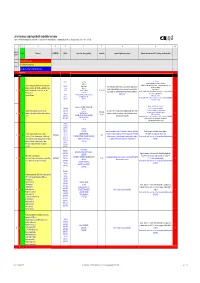
List of Substances Subject to Prohibited / Undesirable / Declaration
List of substances subject to prohibited / undesirable / declaration Appendix 1 to the Guidelines to Design and Purchasing Prohibition of Harmful Substances : ebmpapstMulfingen: 90-001 ebmpapstSt.Georgen: WN 3-12.2 ebmpapstLandshut: 60118.00040 (Ver. 5.0-31.07.2008) 1 2 3 4 5 6 7 8 9 10 Consecu N - new Substance EU-INDEX-No.: CAS-No. Source (law, decree, guideline). Hazard/risk example of application/occurrence Remarks and comments (of 0.1% deviating consideration limit) tive No. V = prohibited (verboten) U = undesirable (unerwünscht) D = subject to declaration (deklarationspflichtig) V = prohibited RoHS from 100 ppm 7439-92-1 TRGS 905, Pb-stabilisers and pigments are subject to declaration. GefStoffV, Prohibited: Anhydrite neutral lead carbonate, lead hydrogen carbonate and lead Lead and compounds (red lead oxide, lead sulphate, lead (7446-14-2 ChemVerbotsV, cable coating, heat transfer medium for accumulators, hybrid integrated sulphate as dye pigment hydrogen carbonate, lead carbonate, lead phtalates, lead 1319-46-6 BatterieV, circuits, stabilisers in plastics, vessels and pipes for aggressive liquids, < 0.4 % in batteries 1 V N acetates, lead phosphates, lead stereates , etc) lead 598-63-0 2002/95/EG (RoHS), K3, R 1, R 3 V E F according to RoHS: Lead as alloying additive in aluminum (up to 0.4 mass%) in steel (up to 67/458/EEC pigment production, lead alloys, anti-corrosion agents (fuel additives), chromate: refer to 0.35 mass%), in copper (up to 4%), 301-04-2 76/769/EEC (89/667/EEC, 97/10/EC, 97/56/EC) soldering agent chromium (VI)