Gene Expression Profiling of Patients with Polycythemia Rubra Vera
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supplementary Materials
Supplementary material. S1. Images from automatic imaging reader Cytation™ 1. A, A’, A’’ present the same field of view. Cells migrating from the upper compartment of the inserts are stained with DID (red color), Cell nuclei are stained with DAPI (blue color). A’ and A’’ demonstrate the method of analysis (A’ – nuclei numbering, A’’ – migrating cells numbering), scale bar – 300 µm. ASC BM-MSC Y BM-MSC A mean SEM mean SEM mean SEM CXCL6 2.240 0.727 4.158 0.677 2.149 1.816 CXCL16 4.277 0.248 0.763 0.405 1.130 0.346 CXCL12 -2.855 0.483 -4.528 0.226 -3.616 0.318 SMAD3 -0.511 0.191 -1.522 0.188 -1.332 0.215 TGFB2 3.742 0.533 1.238 0.210 0.990 0.568 COL14A 1.532 0.357 -0.397 0.736 -1.522 0.469 MHX 1.397 0.414 1.145 0.412 0.642 0.613 SCX 2.984 0.301 2.062 0.320 2.031 0.249 RUNX2 3.290 0.359 2.319 0.388 2.546 0.529 PPRAG 1.720 0.303 2.926 0.423 2.912 0.215 Supplementary material S3. The table provides the mean Δct and SEM values for all genes and all groups analyzed in this study (n=8 in each group). Supplementary material S2. The table provides the list of genes which displayed significantly different expression between hASCs and hBM-MSCs A based on microarray nr Exp p-value Exp Fold Change ID Symbol Entrez Gene Name 1 2.84E-05 16.896 16960355 TM4SF1 transmembrane 4 L six family member 1 2 9.10E-09 12.238 16684080 IFI6 interferon alpha inducible protein 6 3 3.93E-08 11.851 16667702 VCAM1 vascular cell adhesion molecule 1 4 1.54E-05 9.944 16840113 CXCL16 C-X-C motif chemokine ligand 16 5 2.01E-06 9.123 16852463 RAB27B RAB27B, member RAS oncogene -

Identification of Transcriptional Mechanisms Downstream of Nf1 Gene Defeciency in Malignant Peripheral Nerve Sheath Tumors Daochun Sun Wayne State University
Wayne State University DigitalCommons@WayneState Wayne State University Dissertations 1-1-2012 Identification of transcriptional mechanisms downstream of nf1 gene defeciency in malignant peripheral nerve sheath tumors Daochun Sun Wayne State University, Follow this and additional works at: http://digitalcommons.wayne.edu/oa_dissertations Recommended Citation Sun, Daochun, "Identification of transcriptional mechanisms downstream of nf1 gene defeciency in malignant peripheral nerve sheath tumors" (2012). Wayne State University Dissertations. Paper 558. This Open Access Dissertation is brought to you for free and open access by DigitalCommons@WayneState. It has been accepted for inclusion in Wayne State University Dissertations by an authorized administrator of DigitalCommons@WayneState. IDENTIFICATION OF TRANSCRIPTIONAL MECHANISMS DOWNSTREAM OF NF1 GENE DEFECIENCY IN MALIGNANT PERIPHERAL NERVE SHEATH TUMORS by DAOCHUN SUN DISSERTATION Submitted to the Graduate School of Wayne State University, Detroit, Michigan in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY 2012 MAJOR: MOLECULAR BIOLOGY AND GENETICS Approved by: _______________________________________ Advisor Date _______________________________________ _______________________________________ _______________________________________ © COPYRIGHT BY DAOCHUN SUN 2012 All Rights Reserved DEDICATION This work is dedicated to my parents and my wife Ze Zheng for their continuous support and understanding during the years of my education. I could not achieve my goal without them. ii ACKNOWLEDGMENTS I would like to express tremendous appreciation to my mentor, Dr. Michael Tainsky. His guidance and encouragement throughout this project made this dissertation come true. I would also like to thank my committee members, Dr. Raymond Mattingly and Dr. John Reiners Jr. for their sustained attention to this project during the monthly NF1 group meetings and committee meetings, Dr. -

Qt38n028mr Nosplash A3e1d84
! ""! ACKNOWLEDGEMENTS I dedicate this thesis to my parents who inspired me to become a scientist through invigorating scientific discussions at the dinner table even when I was too young to understand what the hippocampus was. They also prepared me for the ups and downs of science and supported me through all of these experiences. I would like to thank my advisor Dr. Elizabeth Blackburn and my thesis committee members Dr. Eric Verdin, and Dr. Emmanuelle Passegue. Liz created a nurturing and supportive environment for me to explore my own ideas, while at the same time teaching me how to love science, test my questions, and of course provide endless ways to think about telomeres and telomerase. Eric and Emmanuelle both gave specific critical advice about the proper experiments for T cells and both volunteered their lab members for further critical advice. I always felt inspired with a sense of direction after thesis committee meetings. The Blackburn lab is full of smart and dedicated scientists whom I am thankful for their support. Specifically Dr. Shang Li and Dr. Brad Stohr for their stimulating scientific debates and “arguments.” Dr. Jue Lin, Dana Smith, Kyle Lapham, Dr. Tet Matsuguchi, and Kyle Jay for their friendships and discussions about what my data could possibly mean. Dr. Eva Samal for teaching me molecular biology techniques and putting up with my late night lab exercises. Beth Cimini for her expertise with microscopy, FACs, singing, and most of all for being a caring and supportive friend. Finally, I would like to thank Dr. Imke Listerman, my scientific partner for most of the breast cancer experiments. -

Solute Carrier Transporters (Slcs) As Possible Drug Targets for Cystic Fibrosis
UNIVERSIDADE DE LISBOA FACULDADE DE CIÊNCIAS DEPARTAMENTO DE QUÍMICA E BIOQUÍMICA Solute Carrier Transporters (SLCs) as Possible Drug Targets for Cystic Fibrosis Íris Lameiro Petinga Mestrado em Bioquímica Especialização em Bioquímica Médica Dissertação orientada por: Professora Doutora Margarida D. Amaral 2017 Acknowledgments/Agradecimentos Ao concluir este trabalho não posso deixar de agradecer a todas as pessoas que contribuíram de alguma maneira para a sua realização. Em primeiro lugar gostaria de agradecer à professora Margarida Amaral não só por me ter recebido no seu laboratório e grupo de investigação, mas também por toda a orientação e acompanhamento e acima de tudo confiança depositada. Não podia deixar também de agradecer ao professor Carlos Farinha por se ter mostrado sempre disponível para me ajudar, por toda a atenção, paciência e por me ter cativado para a área do estudo da Fibrose Quística desde muito cedo. Agradeço a todos os meus colegas de laboratório que sempre se mostraram disponíveis para me ajudar, todos eles à sua maneira. Primeiramente gostaria de agradecer à Verónica que tornou todos os infindáveis “Colony PCR” mais divertidos e me ajudou a seguir em frente quando tudo corria mal. À Sara cuja paciente infinitiva em me ensinar a trabalhar na cultura e grande parte das outras técnicas que utilizei neste trabalho, para além de responder sempre com boa disposição a todas as minhas dúvidas e questões que foram enumeras e muitas delas sem nexo, por vezes. À Madalena e aos seus maravilhosos cadernos de laboratório, sem dúvida a minha fonte de inspiração, mesmo longe esteve sempre disponível para responder às minhas dúvidas por mais chatas e aborrecidas que fossem e me fez crer desde o primeiro dia que seria possível. -
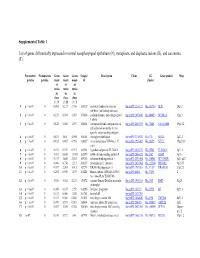
Supplemental Table 1 List of Genes Differentially Expressed In
Supplemental Table 1 List of genes differentially expressed in normal nasopharyngeal epithelium (N), metaplastic and displastic lesions (R), and carcinoma (T). Parametric Permutation Geom Geom Geom Unique Description Clone UG Gene symbol Map p-value p-value mean mean mean id cluster of of of ratios ratios ratios in in in class class class 1 : N 2 : R 3 : T 1 p < 1e-07 0 0.061 0.123 2.708 169329 secretory leukocyte protease IncytePD:2510171 Hs.251754 SLPI 20q12 inhibitor (antileukoproteinase) 2 p < 1e-07 0 0.125 0.394 1.863 163628 sodium channel, nonvoltage-gated IncytePD:1453049 Hs.446415 SCNN1A 12p13 1 alpha 3 p < 1e-07 0 0.122 0.046 1.497 160401 carcinoembryonic antigen-related IncytePD:2060355 Hs.73848 CEACAM6 19q13.2 cell adhesion molecule 6 (non- specific cross reacting antigen) 4 p < 1e-07 0 0.675 1.64 5.594 165101 monoglyceride lipase IncytePD:2174920 Hs.6721 MGLL 3q21.3 5 p < 1e-07 0 0.182 0.487 0.998 166827 nei endonuclease VIII-like 1 (E. IncytePD:1926409 Hs.28355 NEIL1 15q22.33 coli) 6 p < 1e-07 0 0.194 0.339 0.915 162931 hypothetical protein FLJ22418 IncytePD:2816379 Hs.36563 FLJ22418 1p11.1 7 p < 1e-07 0 1.313 0.645 13.593 162399 S100 calcium binding protein P IncytePD:2060823 Hs.2962 S100P 4p16 8 p < 1e-07 0 0.157 1.445 2.563 169315 selenium binding protein 1 IncytePD:2591494 Hs.334841 SELENBP1 1q21-q22 9 p < 1e-07 0 0.046 0.738 1.213 160115 prominin-like 1 (mouse) IncytePD:2070568 Hs.112360 PROML1 4p15.33 10 p < 1e-07 0 0.787 2.264 3.013 167294 HRAS-like suppressor 3 IncytePD:1969263 Hs.37189 HRASLS3 11q12.3 11 p < 1e-07 0 0.292 0.539 1.493 168221 Homo sapiens cDNA FLJ13510 IncytePD:64451 Hs.37896 2 fis, clone PLACE1005146. -

4211C3bea57068cc0a32976fb2
International Journal of Molecular Sciences Article Multiple Known Mechanisms and a Possible Role of an Enhanced Immune System in Bt-Resistance in a Field Population of the Bollworm, Helicoverpa zea: Differences in Gene Expression with RNAseq Roger D. Lawrie 1,2 , Robert D. Mitchell III 3, Jean Marcel Deguenon 2, Loganathan Ponnusamy 2 , Dominic Reisig 4, Alejandro Del Pozo-Valdivia 4, Ryan W. Kurtz 5 and R. Michael Roe 1,2,* 1 Department of Biology/Environmental and Molecular Toxicology Program, 850 Main Campus Dr, North Carolina State University, Raleigh, NC 27695, USA; [email protected] 2 Department of Entomology and Plant Pathology, Campus Box 7647, 3230 Ligon Street, North Carolina State University, Raleigh, NC 27695, USA; [email protected] (J.M.D.); [email protected] (L.P.) 3 Knipling-Bushland US Livestock Insects Research Laboratory Genomics Center, 2700 Fredericksburg Road, United States Department of Agriculture-Agricultural Research Service, Kerrville, TX 78028, USA; [email protected] 4 Department of Entomology and Plant Pathology, Vernon G. James Research & Extension Center, 207 Research Station Road, Plymouth, NC 27962, USA; [email protected] (D.R.); [email protected] (A.D.P.-V.) 5 Cotton Incorporated, 6399 Weston Parkway, Cary, NC 27513, USA; [email protected] * Correspondence: [email protected]; Tel.: +1-919-515-4325 Received: 31 July 2020; Accepted: 1 September 2020; Published: 7 September 2020 Abstract: Several different agricultural insect pests have developed field resistance to Bt (Bacillus thuringiensis) proteins (ex. Cry1Ac, Cry1F, etc.) expressed in crops, including corn and cotton. In the bollworm, Helicoverpa zea, resistance levels are increasing; recent reports in 2019 show up to 1000-fold levels of resistance to Cry1Ac, a major insecticidal protein in Bt-crops. -

Analysis of the Impact of Pufas on Placental Gene Expression V2.28 Uploadx
TECHNISCHE UNIVERSITÄT MÜNCHEN Lehrstuhl für Ernährungsmedizin Analysis of the Impact of Polyunsaturated Fatty Acids (PUFAs) on Placental Gene Expression Eva-Maria Sedlmeier Vollständiger Abdruck der von der Fakultät Wissenschaftszentrum Weihenstephan für Ernährung, Landnutzung und Umwelt der Technischen Universität München zur Erlangung des akademischen Grades eines Doktors der Naturwissenschaften genehmigten Dissertation. Vorsitzender: Univ.-Prof. Dr. M. Klingenspor Prüfer der Dissertation: 1. Univ.-Prof. Dr. J. J. Hauner 2. Univ.-Prof. Dr. H. Daniel Die Dissertation wurde am 17.10.2013 bei der Technischen Universität München eingereicht und durch die Fakultät Wissenschaftszentrum Weihenstephan für Ernährung, Landnutzung und Umwelt am 12.12.2013 angenommen. Table of contents Table of contents Table of contents ....................................................................................................... I Summary .................................................................................................................. VI Zusammenfassung ................................................................................................ VIII 1. Introduction ........................................................................................................ 1 1.1. Fetal programming - a strategy to prevent the obesity epidemic? ...........................................1 1.1.1. The obesity epidemic ........................................................................................................1 1.1.2. Fetal programming -

1 Novel Expression Signatures Identified by Transcriptional Analysis
ARD Online First, published on October 8, 2009 as 10.1136/ard.2009.108043 Ann Rheum Dis: first published as 10.1136/ard.2009.108043 on 7 October 2009. Downloaded from Novel expression signatures identified by transcriptional analysis of separated leukocyte subsets in SLE and vasculitis 1Paul A Lyons, 1Eoin F McKinney, 1Tim F Rayner, 1Alexander Hatton, 1Hayley B Woffendin, 1Maria Koukoulaki, 2Thomas C Freeman, 1David RW Jayne, 1Afzal N Chaudhry, and 1Kenneth GC Smith. 1Cambridge Institute for Medical Research and Department of Medicine, Addenbrooke’s Hospital, Hills Road, Cambridge, CB2 0XY, UK 2Roslin Institute, University of Edinburgh, Roslin, Midlothian, EH25 9PS, UK Correspondence should be addressed to Dr Paul Lyons or Prof Kenneth Smith, Department of Medicine, Cambridge Institute for Medical Research, Addenbrooke’s Hospital, Hills Road, Cambridge, CB2 0XY, UK. Telephone: +44 1223 762642, Fax: +44 1223 762640, E-mail: [email protected] or [email protected] Key words: Gene expression, autoimmune disease, SLE, vasculitis Word count: 2,906 The Corresponding Author has the right to grant on behalf of all authors and does grant on behalf of all authors, an exclusive licence (or non-exclusive for government employees) on a worldwide basis to the BMJ Publishing Group Ltd and its Licensees to permit this article (if accepted) to be published in Annals of the Rheumatic Diseases and any other BMJPGL products to exploit all subsidiary rights, as set out in their licence (http://ard.bmj.com/ifora/licence.pdf). http://ard.bmj.com/ on October 2, 2021 by guest. Protected copyright. 1 Copyright Article author (or their employer) 2009. -

Downloaded from the Mouse Lysosome Gene Database, Mlgdb
1 Supplemental Figure Legends 2 3 Supplemental Figure S1: Epidermal-specific mTORC1 gain-of-function models show 4 increased mTORC1 activation and down-regulate EGFR and HER2 protein expression in a 5 mTORC1-sensitive manner. (A) Immunoblotting of Rheb1 S16H flox/flox keratinocyte cultures 6 infected with empty or adenoviral cre recombinase for markers of mTORC1 (p-S6, p-4E-BP1) 7 activity. (B) Tsc1 cKO epidermal lysates also show decreased expression of TSC2 by 8 immunoblotting of the same experiment as in Figure 2A. (C) Immunoblotting of Tsc2 flox/flox 9 keratinocyte cultures infected with empty or adenoviral cre recombinase showing decreased EGFR 10 and HER2 protein expression. (D) Expression of EGFR and HER2 was decreased in Tsc1 cre 11 keratinocytes compared to empty controls, and up-regulated in response to Torin1 (1µM, 24 hrs), 12 by immunoblot analyses. Immunoblots are contemporaneous and parallel from the same biological 13 replicate and represent the same experiment as depicted in Figure 7B. (E) Densitometry 14 quantification of representative immunoblot experiments shown in Figures 2E and S1D (r≥3; error 15 bars represent STDEV; p-values by Student’s T-test). 16 17 18 19 20 21 22 23 Supplemental Figure S2: EGFR and HER2 transcription are unchanged with epidermal/ 24 keratinocyte Tsc1 or Rptor loss. Egfr and Her2 mRNA levels in (A) Tsc1 cKO epidermal lysates, 25 (B) Tsc1 cKO keratinocyte lysates and(C) Tsc1 cre keratinocyte lysates are minimally altered 26 compared to their respective controls. (r≥3; error bars represent STDEV; p-values by Student’s T- 27 test). -

Analysis of the Expression and Single-Nucleotide Variant Frequencies of the Buyrophilin-Like 2 Gene in Patients with Uveal Melanoma
Supplementary Online Content Amaro A, Parodi F, Diedrich K, et al. Analysis of the expression and single-nucleotide variant frequencies of the buyrophilin-like 2 gene in patients with uveal melanoma. JAMA Ophthalmol. Published online August 11, 2016. doi: 10.1001/jamaophthalmol.2016.2691. eTable 1. M1- and M2-poloarization genes eTable 2. M1- and M2-polarization genes that are associated with disease-free survival of patients with uveal melanoma This supplementary material has been provided by the authors to give readers additional information about their work. © 2016 American Medical Association. All rights reserved. Downloaded From: https://jamanetwork.com/ on 09/30/2021 eTable 1. M1- and M2-poloarization genes 203308_x NM_000195 | HPS1 3257 Hs.40456 203308_x 10 GO:00070 - AI185798 Hermansky-Pudlak syndrome 1 chr10:100 1E+08 1E+08 -1.13131 6.095084 0.859709 0.14105 1.819564 205022_s NM_001085471 FOXN3 1112 Hs.43428 205022_s 14 GO:00000 - NM_0051 forkhead box N3 chr14:896 89622516 90085494 -0.83339 6.106332 1.983495 0.324826 3.850649 204226_a NM_001164380 STAU2 27067 Hs.56181 204226_a 8 GO:00068 - NM_0143 staufen, RNA binding protein, chr8:7446 74461843 74659943 -0.95841 6.106973 0.850648 0.139291 1.804766 208639_x NM_005742 | PDIA6 10130 Hs.21210 208639_x 2 GO:00064 - BC001312 protein disulfide isomerase chr2:1092 10923518 10952960 -0.37258 6.110976 1.043652 0.170783 2.061971 204295_a NA NA NA NA 204295_a NA NA NA NM_0031 NA chrNA:-1-- -1 -1 -0.94773 6.113623 0.686679 0.112319 1.613194 202609_a NM_004447 | EPS8 2059 Hs.59116 202609_a -

Mouse Zfp266 Conditional Knockout Project (CRISPR/Cas9)
https://www.alphaknockout.com Mouse Zfp266 Conditional Knockout Project (CRISPR/Cas9) Objective: To create a Zfp266 conditional knockout Mouse model (C57BL/6J) by CRISPR/Cas-mediated genome engineering. Strategy summary: The Zfp266 gene (NCBI Reference Sequence: NM_001082485 ; Ensembl: ENSMUSG00000060510 ) is located on Mouse chromosome 9. 9 exons are identified, with the ATG start codon in exon 4 and the TAG stop codon in exon 9 (Transcript: ENSMUST00000174462). Exon 6 will be selected as conditional knockout region (cKO region). Deletion of this region should result in the loss of function of the Mouse Zfp266 gene. To engineer the targeting vector, homologous arms and cKO region will be generated by PCR using BAC clone RP23-82H7 as template. Cas9, gRNA and targeting vector will be co-injected into fertilized eggs for cKO Mouse production. The pups will be genotyped by PCR followed by sequencing analysis. Note: Exon 6 starts from about 6.35% of the coding region. The knockout of Exon 6 will result in frameshift of the gene. The size of intron 5 for 5'-loxP site insertion: 798 bp, and the size of intron 6 for 3'-loxP site insertion: 2930 bp. The size of effective cKO region: ~627 bp. The cKO region does not have any other known gene. Page 1 of 8 https://www.alphaknockout.com Overview of the Targeting Strategy Wildtype allele gRNA region 5' gRNA region 3' 1 4 5 6 9 Targeting vector Targeted allele Constitutive KO allele (After Cre recombination) Legends Exon of mouse Zfp266 Homology arm cKO region loxP site Page 2 of 8 https://www.alphaknockout.com Overview of the Dot Plot Window size: 10 bp Forward Reverse Complement Sequence 12 Note: The sequence of homologous arms and cKO region is aligned with itself to determine if there are tandem repeats. -
Sheet1 Page 1 Gene Symbol Gene Description Entrez Gene ID
Sheet1 RefSeq ID ProbeSets Gene Symbol Gene Description Entrez Gene ID Sequence annotation Seed matches location(s) Ago-2 binding specific enrichment (replicate 1) Ago-2 binding specific enrichment (replicate 2) OE lysate log2 fold change (replicate 1) OE lysate log2 fold change (replicate 2) Probability Pulled down in Karginov? NM_005646 202813_at TARBP1 Homo sapiens TAR (HIV-1) RNA binding protein 1 (TARBP1), mRNA. 6894 TR(1..5130)CDS(1..4866) 4868..4874,5006..5013 3.73 2.53 -1.54 -0.44 1 Yes NM_001665 203175_at RHOG Homo sapiens ras homolog gene family, member G (rho G) (RHOG), mRNA. 391 TR(1..1332)CDS(159..734) 810..817,782..788,790..796,873..879 3.56 2.78 -1.62 -1 1 Yes NM_002742 205880_at PRKD1 Homo sapiens protein kinase D1 (PRKD1), mRNA. 5587 TR(1..3679)CDS(182..2920) 3538..3544,3202..3208 4.15 1.83 -2.55 -0.42 1 Yes NM_003068 213139_at SNAI2 Homo sapiens snail homolog 2 (Drosophila) (SNAI2), mRNA. 6591 TR(1..2101)CDS(165..971) 1410..1417,1814..1820,1610..1616 3.5 2.79 -1.38 -0.31 1 Yes NM_006270 212647_at RRAS Homo sapiens related RAS viral (r-ras) oncogene homolog (RRAS), mRNA. 6237 TR(1..1013)CDS(46..702) 871..877 3.82 2.27 -1.54 -0.55 1 Yes NM_025188 219923_at,242056_at TRIM45 Homo sapiens tripartite motif-containing 45 (TRIM45), mRNA. 80263 TR(1..3584)CDS(589..2331) 3408..3414,2437..2444,3425..3431,2781..2787 3.87 1.89 -0.62 -0.09 1 Yes NM_024684 221600_s_at,221599_at C11orf67 Homo sapiens chromosome 11 open reading frame 67 (C11orf67), mRNA.