SCIENTIFIC REVIEWS Predicting 3D Protein Structure by Computational Approach from Amino Acid Sequence a Leap Forward in Solving the Classic Problem of Biochemistry
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Cambridge's 92 Nobel Prize Winners Part 2 - 1951 to 1974: from Crick and Watson to Dorothy Hodgkin
Cambridge's 92 Nobel Prize winners part 2 - 1951 to 1974: from Crick and Watson to Dorothy Hodgkin By Cambridge News | Posted: January 18, 2016 By Adam Care The News has been rounding up all of Cambridge's 92 Nobel Laureates, celebrating over 100 years of scientific and social innovation. ADVERTISING In this installment we move from 1951 to 1974, a period which saw a host of dramatic breakthroughs, in biology, atomic science, the discovery of pulsars and theories of global trade. It's also a period which saw The Eagle pub come to national prominence and the appearance of the first female name in Cambridge University's long Nobel history. The Gender Pay Gap Sale! Shop Online to get 13.9% off From 8 - 11 March, get 13.9% off 1,000s of items, it highlights the pay gap between men & women in the UK. Shop the Gender Pay Gap Sale – now. Promoted by Oxfam 1. 1951 Ernest Walton, Trinity College: Nobel Prize in Physics, for using accelerated particles to study atomic nuclei 2. 1951 John Cockcroft, St John's / Churchill Colleges: Nobel Prize in Physics, for using accelerated particles to study atomic nuclei Walton and Cockcroft shared the 1951 physics prize after they famously 'split the atom' in Cambridge 1932, ushering in the nuclear age with their particle accelerator, the Cockcroft-Walton generator. In later years Walton returned to his native Ireland, as a fellow of Trinity College Dublin, while in 1951 Cockcroft became the first master of Churchill College, where he died 16 years later. 3. 1952 Archer Martin, Peterhouse: Nobel Prize in Chemistry, for developing partition chromatography 4. -

Crystal Vision Dorothy Hodgkin & the Nobel Prize “Few Were Her Equal in Generosity of Spirit, Breadth of Mind, Cultivated Humaneness, Or Gift for Giving
Crystal Vision Dorothy Hodgkin & the Nobel Prize “Few were her equal in generosity of spirit, breadth of mind, cultivated humaneness, or gift for giving. She should be remembered not only for a lifetime’s succession of brilliantly achieved structures. While those who knew her, experienced her quiet and modest and extremely powerful influence, learned from her more than the positioning of atoms in the three-dimensional molecule, she will be remembered not only with respect, and reverence, and gratitude, but more than anything else, with love. Let that be her lasting memorial.” Anne Sayre, in the Autumn 1995 Newsletter of the American Crystallographic Association Cover image © Emilio Segre Visual Archives/American Institute of Physics/Science Photo Library Letter from the Principal Dr Alice Prochaska, Somerville College Many remarkable woman scientists have passed through Somerville, but few rival Dorothy Hodgkin, whose path-breaking work in crystallography showed a mind not only attuned to high science but one also able to envision a crystal structure from the pattern made by its X-ray diffraction. Her ability to ‘see’ molecules such as cholesterol, penicillin, vitamin B and insulin transformed her field. 2014 sees the fiftieth anniversary of Professor Hodgkin’s Nobel prize. It is also the International Year of Crystallography, marking the 100th anniversary of Max von Laue’s Nobel Prize for Physics, awarded for his discovery that X-rays could be diffracted by crystals. That discovery underpinned Dorothy Hodgkin’s work. We are proud that Somerville supported her at a time when there was widespread opposition to married women pursuing academic careers. -
RSC Branding
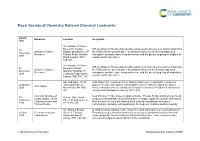
Royal Society of Chemistry National Chemical Landmarks Award Honouree Location Inscription Date The Institute of Cancer Research, Chester ICR scientists on this site and elsewhere pioneered numerous new cancer drugs from 10 Institute of Cancer Beatty Laboratories, 237 the 1950s until the present day – including the discovery of chemotherapy drug December Research Fulham Road, Chelsea carboplatin, prostate cancer drug abiraterone and the genetic targeting of olaparib for 2018 Road, London, SW3 ovarian and breast cancer. 6JB, UK The Institute of Cancer ICR scientists on this site and elsewhere pioneered numerous new cancer drugs from 10 Research, Royal Institute of Cancer the 1950s until the present day – including the discovery of chemotherapy drug December Marsden Hospital, 15 Research carboplatin, prostate cancer drug abiraterone and the genetic targeting of olaparib for 2018 Cotswold Road, Sutton, ovarian and breast cancer. London, SM2 5NG, UK Ape and Apple, 28-30 John Dalton Street was opened in 1846 by Manchester Corporation in honour of 26 October John Dalton Street, famous chemist, John Dalton, who in Manchester in 1803 developed the Atomic John Dalton 2016 Manchester, M2 6HQ, Theory which became the foundation of modern chemistry. President of Manchester UK Literary and Philosophical Society 1816-1844. Chemical structure of Near this site in 1903, James Colquhoun Irvine, Thomas Purdie and their team found 30 College Gate, North simple sugars, James a way to understand the chemical structure of simple sugars like glucose and lactose. September Street, St Andrews, Fife, Colquhoun Irvine and Over the next 18 years this allowed them to lay the foundations of modern 2016 KY16 9AJ, UK Thomas Purdie carbohydrate chemistry, with implications for medicine, nutrition and biochemistry. -

Robert Burns Woodward
The Life and Achievements of Robert Burns Woodward Long Literature Seminar July 13, 2009 Erika A. Crane “The structure known, but not yet accessible by synthesis, is to the chemist what the unclimbed mountain, the uncharted sea, the untilled field, the unreached planet, are to other men. The achievement of the objective in itself cannot but thrill all chemists, who even before they know the details of the journey can apprehend from their own experience the joys and elations, the disappointments and false hopes, the obstacles overcome, the frustrations subdued, which they experienced who traversed a road to the goal. The unique challenge which chemical synthesis provides for the creative imagination and the skilled hand ensures that it will endure as long as men write books, paint pictures, and fashion things which are beautiful, or practical, or both.” “Art and Science in the Synthesis of Organic Compounds: Retrospect and Prospect,” in Pointers and Pathways in Research (Bombay:CIBA of India, 1963). Robert Burns Woodward • Graduated from MIT with his Ph.D. in chemistry at the age of 20 Woodward taught by example and captivated • A tenured professor at Harvard by the age of 29 the young... “Woodward largely taught principles and values. He showed us by • Published 196 papers before his death at age example and precept that if anything is worth 62 doing, it should be done intelligently, intensely • Received 24 honorary degrees and passionately.” • Received 26 medals & awards including the -Daniel Kemp National Medal of Science in 1964, the Nobel Prize in 1965, and he was one of the first recipients of the Arthur C. -

Max Perutz (1914–2002)
PERSONAL NEWS NEWS Max Perutz (1914–2002) Max Perutz died on 6 February 2002. He Nobel Prize for Chemistry in 1962 with structure is more relevant now than ever won the Nobel Prize for Chemistry in his colleague and his first student John as we turn attention to the smallest 1962 after determining the molecular Kendrew for their work on the structure building blocks of life to make sense of structure of haemoglobin, the red protein of haemoglobin (Perutz) and myoglobin the human genome and mechanisms of in blood that carries oxygen from the (Kendrew). He was one of the greatest disease.’ lungs to the body tissues. Perutz attemp- ambassadors of science, scientific method Perutz described his work thus: ted to understand the riddle of life in the and philosophy. Apart from being a great ‘Between September 1936 and May 1937 structure of proteins and peptides. He scientist, he was a very kindly and Zwicky took 300 or more photographs in founded one of Britain’s most successful tolerant person who loved young people which he scanned between 5000 and research institutes, the Medical Research and was passionately committed towards 10,000 nebular images for new stars. Council Laboratory of Molecular Bio- societal problems, social justice and This led him to the discovery of one logy (LMB) in Cambridge. intellectual honesty. His passion was to supernova, revealing the final dramatic Max Perutz was born in Vienna in communicate science to the public and moment in the death of a star. Zwicky 1914. He came from a family of textile he continuously lectured to scientists could say, like Ferdinand in The Tempest manufacturers and went to the Theresium both young and old, in schools, colleges, when he had to hew wood: School, named after Empress Maria universities and research institutes. -

Guides to the Royal Institution of Great Britain: 1 HISTORY
Guides to the Royal Institution of Great Britain: 1 HISTORY Theo James presenting a bouquet to HM The Queen on the occasion of her bicentenary visit, 7 December 1999. by Frank A.J.L. James The Director, Susan Greenfield, looks on Front page: Façade of the Royal Institution added in 1837. Watercolour by T.H. Shepherd or more than two hundred years the Royal Institution of Great The Royal Institution was founded at a meeting on 7 March 1799 at FBritain has been at the centre of scientific research and the the Soho Square house of the President of the Royal Society, Joseph popularisation of science in this country. Within its walls some of the Banks (1743-1820). A list of fifty-eight names was read of gentlemen major scientific discoveries of the last two centuries have been made. who had agreed to contribute fifty guineas each to be a Proprietor of Chemists and physicists - such as Humphry Davy, Michael Faraday, a new John Tyndall, James Dewar, Lord Rayleigh, William Henry Bragg, INSTITUTION FOR DIFFUSING THE KNOWLEDGE, AND FACILITATING Henry Dale, Eric Rideal, William Lawrence Bragg and George Porter THE GENERAL INTRODUCTION, OF USEFUL MECHANICAL - carried out much of their major research here. The technological INVENTIONS AND IMPROVEMENTS; AND FOR TEACHING, BY COURSES applications of some of this research has transformed the way we OF PHILOSOPHICAL LECTURES AND EXPERIMENTS, THE APPLICATION live. Furthermore, most of these scientists were first rate OF SCIENCE TO THE COMMON PURPOSES OF LIFE. communicators who were able to inspire their audiences with an appreciation of science. -

Honorary Graduates – 1966 - 2004
THE UNIVERSITY OF YORK HONORARY GRADUATES – 1966 - 2004 1966 MR PATRICK BLACKETT, physicist THE RT HON LORD GARDINER, Lord Chancellor SIR PETER HALL, director of plays and operas PRESIDENT KAUNDA, Head of State SIR HENRY MOORE, sculptor SIR ROBERT READ, poet and critic THE RT HON LORD ROBBINS, economist SIR MICHAEL TIPPETT, musician and composer THE RT HON BARONESS WOOTTON OF ABINGER 1967 SIR JOHN DUNNINGTON-JEFFERSON, for services to the University DR ARTHUR GLADWIN. For services to the University PROFESSOR F R LEAVIS, literary critic PROFESSOR WASSILY LEONTIEFF, economist PROFESSOR NIKLAUS PEVSNER, art & architecture critic and historian 1968 AMADEUS QUARTET NORBET BRAININ MARTIN LOVELL SIGMUND NISSELL PETER SCHIDLOF PROFESSOR F W BROOKS, historian LORD CLARK, art historian MRS B PAGE, librarian LORD SWANN, Biologist (and Director-General of the BBC) DAME EILEEN YOUNGHUSBAND, social administrator 1969 PROFESSOR L C KNIGHTS, literacy critic SIR PETER PEARS, singer SIR GEORGE PICKERING, scholar in medicine SIR GEORGE RUSSELL, industrial designer PROFESSOR FRANCIS WORMALD, historian 1969 Chancellor’s installation ceremony THE EARL OF CRAWFORD AND BALCARRES, patron of the arts MR JULIAN CAIN, scholar and librarian PROFESSOR DAVID KNOWLES, historian MR WALTER LIPPMAN, writer & journalist 1970 PROFESSOR ROGER BROWN, social psychologist THE RT HON THE VISCOUNT ESHER, PROFESSOR DOROTHY HODGKIN, chemist PROFESSOR A J P TAYLOR, historian 1971 DAME KITTY ANDERSON, headmistress DR AARON COPLAND, composer DR J FOSTER, secretary-General of the Association -

One Million Structures and Counting the Journey, the Insights, and the Future of the CSD
One Million Structures and Counting The journey, the insights, and the future of the CSD Suzanna Ward The Cambridge Crystallographic Data Centre ECM32 – Wednesday 21st August 2019 2 The Cambridge Structural Database (CSD) 1,013,731 ▪ Every published Structures structure An N-heterocycle published produced by a that year ▪ Inc. ASAP & early view chalcogen-bonding ▪ CSD Communications catalyst. Determined Structures at Shandong published ▪ Patents University in China by previously Yao Wang and his ▪ University repositories team. XOPCAJ - The millionth CSD structure. ▪ Every entry enriched and annotated by experts ▪ Discoverability of data and knowledge ▪ Sustainable for over 54 years 3 Inside the CSD Organic ligands Additional data Organic Metal-Organic • Drugs • 10,860 polymorph families 43% 57% • Agrochemicals • 169,218 melting points • At least one transition metal, Pigments • 840,667 crystal colours lanthanide, actinide or any of Al, • Explosives Ga, In, Tl, Ge, Sn, Pb, Sb, Bi, Po • 700,002 crystal shapes • Protein ligands • 23,622 bioactivity details Polymeric: 11% Polymeric: • 9,740 natural source data Not Polymeric Metal-Organic • > 250,000 oxidation states 89% • Metal Organic Frameworks • Models for new catalysts ligand Links/subsets • Porous frameworks for gas s • Drugbank storage • Druglike • Fundamental chemical bonding • MOFs Single Multi ligands • PDB ligands Component Component • PubChem 56% 44% • ChemSpider • Pesticides 4 1965 The sound of music • Film first released in 1965 • Highest grossing film of 1965 • Set in Austria 5 The 1960s Credits: NASA Credits: Thegreenj 6 The vision • Established in 1965 by Olga Kennard • She and J.D. Bernal had a vision that a collective use of data would lead to new knowledge and generate insights J.D. -

JOHN DESMOND BERNAL – the SAGE by William Reville, University College, Cork
JOHN DESMOND BERNAL – THE SAGE By William Reville, University College, Cork. I remember excitedly buying a boxed set of 4 books, Science in History by John D. Bernal (Pelican, 1965), when I was an undergraduate. At the time I was an amateur Marxist and Bernal’s work was an encyclopaedic analysis of science and society from a Marxist point of view. I was delighted to learn that Bernal was an Irishman who had spent a brilliant career at the leading edge of UK science, making many notable contributions. John Desmond Bernal was born in Nenagh, Co. Tipperary in 1901. The Bernals were originally Sephardic Jews who came to Ireland in 1840 from Spain via Amsterdam and London. They converted to Catholicism and John was Jesuit-educated. John enthusiastically supported the Easter Rising and, as a boy, he organised a Society for Perpetual Adoration. He moved away from religion as an adult, becoming an atheist. Bernal showed precocious talent right from the start. At the age of two he was taken by his American mother to see his grandmother in California and he amazed passengers on the steamship by talking in both English and French. In later life at Cambridge his fellow students nicknamed him ‘Sage’ because of his great knowledge. Bernal started as a science undergraduate at Cambridge in 1919 where his studies included mineralogy and the mathematics of symmetry. He gained a research position in 1923 at the Royal Institution in London with William Henry Bragg the eminent crystallographer (one who studies crystalline structures using X-rays). He returned to Cambridge in 1927 as the first lecturer in structural crystallography, fully convinced of the enormous potential of his chosen field to elucidate the structure of the technological and biological worlds. -

Historical Group
Historical Group NEWSLETTER and SUMMARY OF PAPERS No. 67 Winter 2015 Registered Charity No. 207890 COMMITTEE Chairman: Dr J A Hudson ! Dr C Ceci (RSC) Graythwaite, Loweswater, Cockermouth, ! Dr N G Coley (Open University) Cumbria, CA13 0SU ! Dr C J Cooksey (Watford, Hertfordshire) [e-mail [email protected]] ! Prof A T Dronsfield (Swanwick, Secretary: Prof. J. W. Nicholson ! Derbyshire) 52 Buckingham Road, Hampton, Middlesex, ! Prof E Homburg (University of TW12 3JG [e-mail: [email protected]] ! Maastricht) Membership Prof W P Griffith ! Prof F James (Royal Institution) Secretary: Department of Chemistry, Imperial College, ! Dr M Jewess (Harwell, Oxon) London, SW7 2AZ [e-mail [email protected]] ! Dr D Leaback (Biolink Technology) Treasurer: Dr P J T Morris ! Mr P N Reed (Steensbridge, Science Museum, Exhibition Road, South ! Herefordshire) Kensington, London, SW7 2DD ! Dr V Quirke (Oxford Brookes University) [e-mail: [email protected]] !Prof. H. Rzepa (Imperial College) Newsletter Dr A Simmons !Dr A Sella (University College) Editor Epsom Lodge, La Grande Route de St Jean, St John, Jersey, JE3 4FL [e-mail [email protected]] Newsletter Dr G P Moss Production: School of Biological and Chemical Sciences, Queen Mary University of London, Mile End Road, London E1 4NS [e-mail [email protected]] http://www.chem.qmul.ac.uk/rschg/ http://www.rsc.org/membership/networking/interestgroups/historical/index.asp 1 RSC Historical Group Newsletter No. 67 Winter 2015 Contents From the Editor 2 ROYAL SOCIETY OF CHEMISTRY HISTORICAL GROUP NEWS The Life and Work of Sir John Cornforth CBE AC FRS 3 Royal Society of Chemistry News 4 Feedback from the Summer 2014 Newsletter – Alwyn Davies 4 Another Object to Identify – John Nicholson 4 Published Histories of Chemistry Departments in Britain and Ireland – Bill Griffith 5 SOCIETY NEWS News from the Historical Division of the German Chemical Society – W.H. -

The Royal Society of Chemistry Presidents 1841 T0 2021
The Presidents of the Chemical Society & Royal Society of Chemistry (1841–2024) Contents Introduction 04 Chemical Society Presidents (1841–1980) 07 Royal Society of Chemistry Presidents (1980–2024) 34 Researching Past Presidents 45 Presidents by Date 47 Cover images (left to right): Professor Thomas Graham; Sir Ewart Ray Herbert Jones; Professor Lesley Yellowlees; The President’s Badge of Office Introduction On Tuesday 23 February 1841, a meeting was convened by Robert Warington that resolved to form a society of members interested in the advancement of chemistry. On 30 March, the 77 men who’d already leant their support met at what would be the Chemical Society’s first official meeting; at that meeting, Thomas Graham was unanimously elected to be the Society’s first president. The other main decision made at the 30 March meeting was on the system by which the Chemical Society would be organised: “That the ordinary members shall elect out of their own body, by ballot, a President, four Vice-Presidents, a Treasurer, two Secretaries, and a Council of twelve, four of Introduction whom may be non-resident, by whom the business of the Society shall be conducted.” At the first Annual General Meeting the following year, in March 1842, the Bye Laws were formally enshrined, and the ‘Duty of the President’ was stated: “To preside at all Meetings of the Society and Council. To take the Chair at all ordinary Meetings of the Society, at eight o’clock precisely, and to regulate the order of the proceedings. A Member shall not be eligible as President of the Society for more than two years in succession, but shall be re-eligible after the lapse of one year.” Little has changed in the way presidents are elected; they still have to be a member of the Society and are elected by other members. -

Guide to the Jack Dunitz Papers, 1927-2009 Page 6 of 31
Guide to The Jack Dunitz Papers, 1927-2009 Page 6 of 31 Series Outline 1. Correspondence and Non-Dunitz Publications, 1932-2009 undated 2. Biographical Materials and Photographs, 1967-2008 undated 3. Publications by Jack Dunitz, 1947-2004 4. Manuscripts and Lecture Materials, 1946-2006 undated 5. Research Notes and Notebooks, 1941-1989 6. Subject Files, 1927-2008 Special Collections, The Valley Library PDF Created November 13, 2015 Guide to The Jack Dunitz Papers, 1927-2009 Page 8 of 31 Detailed Description of the Collection Box 1. Correspondence and Non-Dunitz Publications, 1932-2009 undated (26 boxes) The folders in this series contain correspondence and publications by specific authors, as maintained by Dunitz. The correspondence materials include traditional letters as well as a large volume of emails and word processing files printed out over time by Dunitz. Highlights of the series include lengthy exchanges with a number of notable scientists including Carleton Gajdusek, Dorothy Hodgkin and Kenneth Trueblood. The activities of the Cambridge Crystallographic Data Centre are also well-documented in this series, as is Dunitz©s work in preparing his Biographical Memoirs of the Royal Society article on Linus Pauling. Access to the Cambridge Crystallographic Data Centre and the Royal Society correspondence in Series 1 is restricted due to the presence of confidential information. All requests for access to this material should be directed to the University Archivist. 1.001 Correspondence: A, 1976-2008 undated (16 folders) Folder 1.1 Abir-Am,