Neurofibromatosis: an Update of Ophthalmic Characteristics and Applications of Optical Coherence Tomography
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Drug Mechanisms
REVIEW OF OPTOMETRY EARN 2 CE CREDITS: Don’t Be Stumped by These Lumps and Bumps, Page 70 ■ VOL. 154 NO. 4 April 15, 2017 www.reviewofoptometry.com TH ■ 10 ANNUAL APRIL 15, 2017 PHARMACEUTICALS REPORT ■ ANNUAL PHARMACEUTICALS REPORT An Insider’s View of DRUG MECHANISMS ■ CE: DIFFERENTIAL DIAGNOSIS OF EYELID LESIONS CE: DIFFERENTIAL DIAGNOSIS OF EYELID LESIONS You can choose agents with greater precision—and evaluate their performance better—when you know what makes them tick. • How Antibiotics Work—and Why They Sometimes Don’t, Page 30 • Glaucoma Therapy: Finding the Right Combination, Page 46 • Anti-inflammatories: Sort Out Your Many Steroids and NSAIDs, Page 40 • Dry Eye: Master the Science Beneath the Surface, Page 56 • Resist the Itch: Managing Allergic Conjunctivitis, Page 64 001_ro0417_fc.indd 1 4/4/17 2:21 PM . rs ke ee S t S up r po fo rtiv Com e. Nature Lovers. Because I know their eyes are prone to discomfort, I prescribe the 1-DAY ACUVUE® MOIST Family. § 88% of all BLINK STABILIZED® Design contact lenses were fi tted in the fi rst attempt, and 99.5% within 2 trial fittings. ** Based on in vitro data. Clinical studies have not been done directly linking differences in lysozyme profi le with specifi c clinical benefi ts. * UV-blocking percentages are based on an average across the wavelength spectrum. † Helps protect against transmission of harmful UV radiation to the cornea and into the eye. ‡ WARNING: UV-absorbing contact lenses are NOT substitutes for protective UV-absorbing eyewear such as UV-absorbing goggles or sunglasses because they do not completely cover the eye and surrounding area. -

The Ophthalmology Examinations Review
The Ophthalm logy Examinations Review Second EditionSecond Edition 7719tp.indd 1 1/4/11 8:13 PM FA B1037 The Ophthalmology Examinations Review This page intentionally left blank BB1037_FM.indd1037_FM.indd vvii 112/24/20102/24/2010 22:31:16:31:16 PPMM The Ophthalm logy Examinations Review Second Edition Tien Yin WONG National University of Singapore, Singapore & University of Melbourne, Australia With Contributions From Chelvin SNG National University Health System, Singapore Laurence LIM Singapore National Eye Centre, Singapore World Scientific NEW JERSEY • LONDON • SINGAPORE • BEIJING • SHANGHAI • HONG KONG • TAIPEI • CHENNAI 7719tp.indd 2 1/4/11 8:13 PM Published by World Scientific Publishing Co. Pte. Ltd. 5 Toh Tuck Link, Singapore 596224 USA office: 27 Warren Street, Suite 401-402, Hackensack, NJ 07601 UK office: 57 Shelton Street, Covent Garden, London WC2H 9HE Library of Congress Cataloging-in-Publication Data Wong, Tien Yin. The ophthalmology examinations review / Tien Yin Wong ; with contributions from Chelvin Sng, Laurence Lim. -- 2nd ed. p. ; cm. Includes index. ISBN-13: 978-981-4304-40-5 (hardcover : alk. paper) ISBN-10: 981-4304-40-9 (hardcover : alk. paper) ISBN-13: 978-981-4304-41-2 (pbk. : alk. paper) ISBN-10: 981-4304-41-7 (pbk. : alk. paper) 1. Ophthalmology--Outlines, syllabi, etc. 2. Ophthalmology--Examinations, questions, etc. I. Sng, Chelvin. II. Lim, Laurence. III. Title. [DNLM: 1. Eye Diseases--Examination Questions. 2. Ophthalmologic Surgical Procedures--Examination Questions. WW 18.2] RE50.W66 2011 617.7--dc22 2010054298 British Library Cataloguing-in-Publication Data A catalogue record for this book is available from the British Library. -

Scholars Journal of Medical Case Reports Neurofibromatosis Type 1
Nurul AM et al.; Sch J Med Case Rep, November 2015; 3(11):1128-1132 Scholars Journal of Medical Case Reports ISSN 2347-6559 (Online) Sch J Med Case Rep 2015; 3(11):1128-1132 ISSN 2347-9507 (Print) ©Scholars Academic and Scientific Publishers (SAS Publishers) (An International Publisher for Academic and Scientific Resources) Neurofibromatosis Type 1 with Optic Nerve Glioma: A Case Report Nurul AM1, Joseph A2 1 4thyear resident, Department of Ophthalmology, Hospital University Kebangsaan Malaysia, Jalan Yaakob Latif, Bandar TunRazak, 56000 Cheras, Wilayah Persekutuan Kuala Lumpur Malaysia. 2 Paediatric Ophthalmology Consultant, Department of Ophthalmology, Hospital Kuala Lumpur, Jalan Pahang, 50586 Kuala Lumpur Malaysia. *Corresponding author Ayn Masnon Email: [email protected] Abstract: Ocular manifestations are among the criteria included in diagnosing Neurofibromatosis Type 1. Ocular assessments in children with NF1 are important as young children may do not complain of visual impairment until it is advanced. Here in we report a case of Neurofibromatosis Type 1 with optic nerve glioma, in which the diagnosis are aided by imaging; namely B scan and MRI. Keywords: Neurofibromatosis Type 1, optic glioma, B scan, MRI. INTRODUCTION Ocular examination shows best corrected Neurofibromatosis type 1 (NF1) is a common vision for both eyes on first presentation were 6/7.5. inherited disorder with an approximate incidence of Refraction showed low refractive error and the child not 1:3000 [1]. It is an autosomal dominant disorder, with requiring glasses. Hirschberg Test was central and no the mutation of a tumor suppressor gene, located on the squint noted. No proptosis (Figure 1) and extra ocular long arm of chromosome 17q11 [2]. -

Uživatel:Zef/Output18
Uživatel:Zef/output18 < Uživatel:Zef rozřadit, rozdělit na více článků/poznávaček; Název !! Klinický obraz !! Choroba !! Autor Bárányho manévr; Bonnetův manévr; Brudzinského manévr; Fournierův manévr; Fromentův manévr; Heimlichův manévr; Jendrassikův manévr; Kernigův manévr; Lasčgueův manévr; Müllerův manévr; Scanzoniho manévr; Schoberův manévr; Stiborův manévr; Thomayerův manévr; Valsalvův manévr; Beckwithova známka; Sehrtova známka; Simonova známka; Svěšnikovova známka; Wydlerova známka; Antonovo znamení; Apleyovo znamení; Battleho znamení; Blumbergovo znamení; Böhlerovo znamení; Courvoisierovo znamení; Cullenovo znamení; Danceovo znamení; Delbetovo znamení; Ewartovo znamení; Forchheimerovo znamení; Gaussovo znamení; Goodellovo znamení; Grey-Turnerovo znamení; Griesingerovo znamení; Guddenovo znamení; Guistovo znamení; Gunnovo znamení; Hertogheovo znamení; Homansovo znamení; Kehrerovo znamení; Leserovo-Trélatovo znamení; Loewenbergerovo znamení; Minorovo znamení; Murphyho znamení; Nobleovo znamení; Payrovo znamení; Pembertonovo znamení; Pinsovo znamení; Pleniesovo znamení; Pléniesovo znamení; Prehnovo znamení; Rovsingovo znamení; Salusovo znamení; Sicardovo znamení; Stellwagovo znamení; Thomayerovo znamení; Wahlovo znamení; Wegnerovo znamení; Zohlenovo znamení; Brachtův hmat; Credého hmat; Dessaignes ; Esmarchův hmat; Fritschův hmat; Hamiltonův hmat; Hippokratův hmat; Kristellerův hmat; Leopoldovy hmat; Lepagův hmat; Pawlikovovy hmat; Riebemontův-; Zangmeisterův hmat; Leopoldovy hmaty; Pawlikovovy hmaty; Hamiltonův znak; Spaldingův znak; -

Table I. Genodermatoses with Known Gene Defects 92 Pulkkinen
92 Pulkkinen, Ringpfeil, and Uitto JAM ACAD DERMATOL JULY 2002 Table I. Genodermatoses with known gene defects Reference Disease Mutated gene* Affected protein/function No.† Epidermal fragility disorders DEB COL7A1 Type VII collagen 6 Junctional EB LAMA3, LAMB3, ␣3, 3, and ␥2 chains of laminin 5, 6 LAMC2, COL17A1 type XVII collagen EB with pyloric atresia ITGA6, ITGB4 ␣64 Integrin 6 EB with muscular dystrophy PLEC1 Plectin 6 EB simplex KRT5, KRT14 Keratins 5 and 14 46 Ectodermal dysplasia with skin fragility PKP1 Plakophilin 1 47 Hailey-Hailey disease ATP2C1 ATP-dependent calcium transporter 13 Keratinization disorders Epidermolytic hyperkeratosis KRT1, KRT10 Keratins 1 and 10 46 Ichthyosis hystrix KRT1 Keratin 1 48 Epidermolytic PPK KRT9 Keratin 9 46 Nonepidermolytic PPK KRT1, KRT16 Keratins 1 and 16 46 Ichthyosis bullosa of Siemens KRT2e Keratin 2e 46 Pachyonychia congenita, types 1 and 2 KRT6a, KRT6b, KRT16, Keratins 6a, 6b, 16, and 17 46 KRT17 White sponge naevus KRT4, KRT13 Keratins 4 and 13 46 X-linked recessive ichthyosis STS Steroid sulfatase 49 Lamellar ichthyosis TGM1 Transglutaminase 1 50 Mutilating keratoderma with ichthyosis LOR Loricrin 10 Vohwinkel’s syndrome GJB2 Connexin 26 12 PPK with deafness GJB2 Connexin 26 12 Erythrokeratodermia variabilis GJB3, GJB4 Connexins 31 and 30.3 12 Darier disease ATP2A2 ATP-dependent calcium 14 transporter Striate PPK DSP, DSG1 Desmoplakin, desmoglein 1 51, 52 Conradi-Hu¨nermann-Happle syndrome EBP Delta 8-delta 7 sterol isomerase 53 (emopamil binding protein) Mal de Meleda ARS SLURP-1 -

Neurocutaneous Syndromes Maria A
6 Neurocutaneous Syndromes Maria A. Musarella he neurocutaneous disorders are a group of clinically and Tgenetically heterogeneous diseases that are characterized mainly by harmatomas and tumor growth, involving tissues derived by the embryonic germ layer. Older literature has called these disorders “phakomatoses” (mother-spot). The modern nomenclature and traditional eponyms of these entities are given in Table 6-1. Each of the neurocutaneous diseases is rec- ognized as a distinct clinical disorder. This chapter covers the ophthalmic aspects of these syn- dromes, as well as the numerous and varied multisystemic man- ifestations. Although Proteus syndrome and multiple endocrine neoplasia (MEN) 2B are considered separate from the neurocu- taneous diseases, they are covered here because of the clinical resemblance to classic phakomatoses. NEUROFIBROMATOSIS 1 Historical Perspective Dr. Robert William Smith first described neurofibromatosis 1 (NF1) in 1849 in his treatise on Pathology Diagnosis and Treat- ment of Neuroma.100 However, this work received little atten- tion. Neurofibromatosis is most closely linked with the German pathologist, von Recklinghausen, who described the main fea- tures of this entity in his classic paper of 1882.111 Etiology About 50% of cases of NF1 result from new mutations. The NF1 gene has been mapped to 17q11.2 and positionally cloned. The 291 292 handbook of pediatric eye and systemic disease TABLE 6-1. Neurocutaneous Syndromes. Modern nomenclature Eponyms Neurofibromatosis 1 von Recklinghausen disease Peripheral neurofibromatosis Neurofibromatosis 2 Central neurofibromatosis Tuberous sclerosis Bourneville disease von Hippel–Lindau Disease Ataxia telangiectasia Louis–Bar syndrome Sturge–Weber Encephalotrigeminal angiomatosis Klippel–Trenaunay Angiosteohypertrophy Wyburn-Mason syndrome Racemose angiomatosis Multiple endocrine neoplasia 2B Mucosal neuroma syndrome (Wagenmann–Froboese) Proteus syndrome NF1 gene is one of the largest genes in which mutations lead to a disease in humans. -

Neurofibromatosis Type 1 Presenting with Ophthalmic Features: a Case Paediatrics Section Series
DOI: 10.7860/JCDR/2016/21041.8780 Case Series Neurofibromatosis Type 1 Presenting with Ophthalmic Features: A Case Paediatrics Section Series GUNJAN JAIN1, VAIBHAV KUMAR JAIN2, INDRA KUMAR SHARMA3, REENA SHARMA4, NEERAJ SARASWAT5 ABSTRACT Neurofibromatosis type 1 (NF-1) is an autosomal dominant disorder involving multiple systems and affects approximately 1 out of 3000 persons. Ocular manifestations are lisch nodules, plexiform neurofibroma, optic pathway gliomas. The proper diagnosis of NF-1 is a crucial task for a clinician due to the various clinical manifestations including vision and life threatening malignancies in few patients, which may arise in the different phases of life. The authors report three cases of NF-1, presenting with ophthalmic symptoms in teenager boys. On further ophthalmic and paediatric evaluation the diagnosis of NF-1 was confirmed on the basis of clinical criteria. This series also describe the abnormal facial features like telecanthus and broad nose which has been reported rarely. Case 1 was kept under regular follow-up and Case 2 and Case 3 were planned for the debulking surgery for plexiform neurofibroma of upper eye lid. A multidisciplinary approach is required to diagnose and treat such patients keeping in mind the myriad of clinical manifestations and life- long follow-up is required. Keywords: Lisch nodule, Plexiform neurofibroma, Telecanthus CASE 1 for such lesions. Patient was treated for allergic conjunctivitis with A 13-year-old male child presented to out patient services topical fluorometholone and carboxy-methylcellulose 0.5%, 4 of Ophthalmology department with the complaints of muddy times per day in both eyes. Patient was kept under observation discolouration and irritation of both eyes. -

It Would Be a New Diagnostic Criteria for Type 1 Neurofibromatosis?
ORIGINAL ARTICLE Choroidal Freckling: It Would be a New Diagnostic Criteria for Type 1 Neurofi bromatosis? Ozer DURSUN1, Ozlem TEZOL2, Erdem DINC3, Mustafa VATANSEVER1, Gülhan TEMEL4, Pınar DURSUN5, Çağlar CITAK6 ABSTRACT Purpose: The aim of this study was to investigate the frequency of choroidal freckling and evaluate the relationship between choroidal freckling (CF) and Lisch nodule in patients with type 1 neurofi bromatosis (NF-1). Materials-Methods: The study included 23 pediatric patients with NF-1 and 24 age- and sex-matched healthy controls. Presence of Lisch nodule was detected by biomicroscopic examination and recorded. The presence of CF was investigated by near-infrared refl ectance imaging after the pupillary dilation with tropicamide 1%. Diagnostic accuracy of CF was evaluated in patients with NF-1. Results: There was no signifi cant difference in age and gender distributions between the patient and control groups (p>0.05, for both parameters). The Lisch nodule was detected in 60.8% of patients, while no Lisch nodule was observed in the control group. The CF frequency was found in 82.6% in the patient group whereas in 4.16% incontrols. The CF frequency was signifi cantly higher in patients with Lisch nodule compared to patients without (p<0.001). The sensitivity and specifi city for CF fi nding were 82.6% and 95.8%, respectively. Conclusion: The present study showed that choroidal abnormalities termed as CF are common in patients with NF-1 and that it would be accepted as a new diagnostic criterion in the following years. However, there is a need for further studies with larger sample size to assess diagnostic accuracy of CD in patients with NF-1. -

MRCPCH Clinical: Short Cases, History Taking and Communication Skills
MRCPCH Clinical: Short Cases, History Taking and Communication Skills Fourth Edition Simon J Bedwani MBBS MRCPCH Consultant Paediatrician Royal Cornwall Hospital, Truro Vanessa Irvine BSc MBBS MRCPCH Consultant Paediatrician Western Sussex Hospitals NHS Foundation Trust, Chichester R Mark Beattie BSc MBBS FRCPCH MRCP Consultant Paediatric Gastroenterologist University Hospital, Southampton 6415_MRCPCH Clinical Short Cases.indb i 09/03/2016 08:58:54 CONTENTS Introduction v Preparing for the Examination vii The MRCPCH Clinical Examination xi Part I History taking and management planning station 1 1 Introduction and suggested approach 3 2 Cases 9 3 Further scenarios 53 Part II The communication skills station 77 4 Introduction and suggested approach 79 5 Cases 87 6 Further scenarios 121 Part III Clinical stations 139 7 Introduction and suggested approach 141 8 Cardiovascular 147 9 Abdominal 201 10 Respiratory 235 11 Neurological/Neurodisability 263 12 Musculoskeletal and ‘Other’ station 319 12.1 Musculoskeletal 322 12.2 Haematology 357 12.3 Dermatology 381 12.4 Endocrinology and growth 393 12.5 Nephrology 435 13 Child development 449 Index 479 6415_MRCPCH Clinical Short Cases.indb iii 09/03/2016 09:00:01 PART II The communication skills station 6415_MRCPCH Clinical Short Cases.indb 77 09/03/2016 09:00:08 4 INTRODUCTION AND SUGGESTED APPROACH This section of the exam is intended to look at how you communicate with patients and/or their parents in your everyday clinical practice. You are awarded marks for both your communication skills and clinical knowledge, and so it is not enough (although is essential) to just be a good listener – your job is to address patient’s and their family’s concerns appropriately and to provide them with reliable, accurate information about the child’s condition and the ongoing management plan. -

Ocular Manifestations in Neurocutaneous Syndromes with Emphasis on Neurofibromatosis – a Descriptive Observational Study
Jebmh.com Original Research Article Ocular Manifestations in Neurocutaneous Syndromes with Emphasis on Neurofibromatosis – A Descriptive Observational Study Sija Sudha1, Deepa Molathe Gopalan2 1 Department of Ophthalmology, Government T.D. Medical College, Alappuzha, Kerala, India. 2 Department of Ophthalmology, Government Medical College, Ernakulam, Kerala, India. ABSTRACT BACKGROUND Neurocutaneous syndromes (NCS) are a group of genetic disorders that produce Corresponding Author: a variety of developmental abnormalities of the eye. Ophthalmic manifestations Dr. Sija Sudha, Pranamam CSM Nagar, Edapazhinji, usually appear early in life and progress with time. The study was conducted to Thiruvananthapuram – 695010, know the prevalence of ocular manifestations in neurocutaneous syndromes with Kerala, India. emphasis on neurofibromatosis. E-mail: [email protected] METHODS DOI: 10.18410/jebmh/2021/100 This study was conducted in ophthalmology department at a tertiary care hospital during a period of 2 years among 30 patients. All phakomatoses referred from How to Cite This Article: other specialty departments for ophthalmological evaluation and cases diagnosed Sudha S, Gopalan DM. Ocular Manifestations in neurocutaneous in ophthalmology department during routine evaluation were included in the study. syndromes with emphasis on neurofibromatosis – a descriptive RESULTS observational study. J Evid Based Med Neurofibromatosis type 1 (NF-1) accounted for most of (66.67 %) the cases Healthc 2021;8(09):512-516. DOI: followed by Sturge Weber syndrome (SWS) (20 %). Majority (55 %) of NF-1 and 10.18410/jebmh/2021/100 83.33 % of SWS and all patients of other phakomatoses were in the age group < 30 yrs. 55 % of NF-1 patients were males. 65 % of NF-1 patients gave positive Submission 11-09-2020, family history. -

Neurofibromatosis Clinics Association
SKELETAL NEUROFIBROMATOSIS: RESPIRATORY A DISTURBING CIRCULATORY AND ELUSIVE DISEASE ENDOCRINE 2 Pediatric Guidelines/Manifestations 4 Clinical Manifestations of Neurofibromatosis Type 1 and Type 2 6 The Orthopedic Manifestations of NF1 8 Ophthalmologic Considerations for Patients with NF1 and 2 10 Neuro Developmental and Psychological Assessment of NF1 12 NF2 Incidence and Molecular Pathophysiology 16 The Otologic Management of NF2 1 A SERIOUS AND MISUNDERSTOOD DISORDER Although neurofibromatosis has been known to the medical community since 1882, there has been widespread misinformation and a lack of precise medical management guidelines available to both physicians and patients until recently. Pittsburgh’s NF Clinic serves as a beacon of information and medical expertise for thousands of families in the region. At its founding, in 1989, access to medical specialists and diagnostic testing and technology was sporadic, at best. In the 15 years since, physicians, geneticists, social workers, diagnostic technicians, nurses and other service providers have focused their clinical efforts and research interests, resulting in the highest possible care and guidance for NF patients and their families. The highly unpredictable nature of NF requires a level of cross-specialty coordination and communication unlike that of most disorders. This brochure serves as a base reference for primary care givers who can provide the first and most important intervention by recognizing the early warning signs that lead to early diagnosis. The American Academy of Pediatricians issued these guidelines so you can give a proper diagnosis. 1 1 PEDIATRIC GUIDELINES FOR NF1 1 MANIFESTATIONS OF NF The American Academy of Pediatrics issued the NF and other major genetic disorders affect 13 following guidelines for the medical supervision million Americans of a child diagnosed with NF1. -
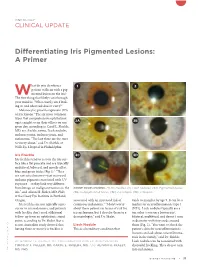
Differentiating Iris Pigmented Lesions: a Primer
ONCOLOGY CLINICAL UPDATE Differentiating Iris Pigmented Lesions: A Primer hat do you do when a 1 2 patient walks in with a pig- Wmented lesion on the iris? The first thing that likely runs through your mind is: “What exactly am I look- ing at, and what risk does it carry?” Melanocytic growths represent 70% of iris lesions.1 The six most common types that comprehensive ophthalmol- ogists might see in their offices on any 3A 3B given day, according to Carol L. Shields, MD, are: freckle, nevus, Lisch nodules, melanocytoma, melanocytosis, and melanoma. “The last three are the ones to worry about,” said Dr. Shields, at Wills Eye Hospital in Philadelphia. Iris Freckle 3C 3D Iris freckles tend to rest on the iris sur- face like a flat pancake and are typically multifocal, bilateral, and mostly affect blue and green irides (Fig 1).2 “They are not actual masses—just increased melanin pigments associated with UV exposure—so they look very different from benign or malignant tumors in the KNOW YOUR LESIONS. (1) Iris freckles. (2) Lisch nodules. (3A) Pigmented nevus, iris,” said Alison H. Skalet, MD, PhD, (3B) nonpigmented nevus, (3C) corectopia, (3D) ectropion. at the Casey Eye Institute in Portland, Oregon. associated with an increased risk of tends to manifest by age 5. It can be a Iris freckles are not typically a pre- cutaneous melanoma.3 “I don’t worry marker for neurofibromatosis type 1 cursor to iris melanoma, and patients about these patients in terms of risk for (NF1). Lisch nodules typically are a with freckles don’t need additional iris melanoma, but I do refer them to a tan color (even on a brown iris), follow-up from an ophthalmic stand- dermatologist,” said Dr.