Hagarman, James.Pdf (9.746Mb)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Title a New Centrosomal Protein Regulates Neurogenesis By
Title A new centrosomal protein regulates neurogenesis by microtubule organization Authors: Germán Camargo Ortega1-3†, Sven Falk1,2†, Pia A. Johansson1,2†, Elise Peyre4, Sanjeeb Kumar Sahu5, Loïc Broic4, Camino De Juan Romero6, Kalina Draganova1,2, Stanislav Vinopal7, Kaviya Chinnappa1‡, Anna Gavranovic1, Tugay Karakaya1, Juliane Merl-Pham8, Arie Geerlof9, Regina Feederle10,11, Wei Shao12,13, Song-Hai Shi12,13, Stefanie M. Hauck8, Frank Bradke7, Victor Borrell6, Vijay K. Tiwari§, Wieland B. Huttner14, Michaela Wilsch- Bräuninger14, Laurent Nguyen4 and Magdalena Götz1,2,11* Affiliations: 1. Institute of Stem Cell Research, Helmholtz Center Munich, German Research Center for Environmental Health, Munich, Germany. 2. Physiological Genomics, Biomedical Center, Ludwig-Maximilian University Munich, Germany. 3. Graduate School of Systemic Neurosciences, Biocenter, Ludwig-Maximilian University Munich, Germany. 4. GIGA-Neurosciences, Molecular regulation of neurogenesis, University of Liège, Belgium 5. Institute of Molecular Biology (IMB), Mainz, Germany. 6. Instituto de Neurociencias, Consejo Superior de Investigaciones Científicas and Universidad Miguel Hernández, Sant Joan d’Alacant, Spain. 7. Laboratory for Axon Growth and Regeneration, German Center for Neurodegenerative Diseases (DZNE), Bonn, Germany. 8. Research Unit Protein Science, Helmholtz Centre Munich, German Research Center for Environmental Health, Munich, Germany. 9. Protein Expression and Purification Facility, Institute of Structural Biology, Helmholtz Center Munich, German Research Center for Environmental Health, Munich, Germany. 10. Institute for Diabetes and Obesity, Monoclonal Antibody Core Facility, Helmholtz Center Munich, German Research Center for Environmental Health, Munich, Germany. 11. SYNERGY, Excellence Cluster of Systems Neurology, Biomedical Center, Ludwig- Maximilian University Munich, Germany. 12. Developmental Biology Program, Sloan Kettering Institute, Memorial Sloan Kettering Cancer Center, New York, USA 13. -

Synergistic Genetic Interactions Between Pkhd1 and Pkd1 Result in an ARPKD-Like Phenotype in Murine Models
BASIC RESEARCH www.jasn.org Synergistic Genetic Interactions between Pkhd1 and Pkd1 Result in an ARPKD-Like Phenotype in Murine Models Rory J. Olson,1 Katharina Hopp ,2 Harrison Wells,3 Jessica M. Smith,3 Jessica Furtado,1,4 Megan M. Constans,3 Diana L. Escobar,3 Aron M. Geurts,5 Vicente E. Torres,3 and Peter C. Harris 1,3 Due to the number of contributing authors, the affiliations are listed at the end of this article. ABSTRACT Background Autosomal recessive polycystic kidney disease (ARPKD) and autosomal dominant polycystic kidney disease (ADPKD) are genetically distinct, with ADPKD usually caused by the genes PKD1 or PKD2 (encoding polycystin-1 and polycystin-2, respectively) and ARPKD caused by PKHD1 (encoding fibrocys- tin/polyductin [FPC]). Primary cilia have been considered central to PKD pathogenesis due to protein localization and common cystic phenotypes in syndromic ciliopathies, but their relevance is questioned in the simple PKDs. ARPKD’s mild phenotype in murine models versus in humans has hampered investi- gating its pathogenesis. Methods To study the interaction between Pkhd1 and Pkd1, including dosage effects on the phenotype, we generated digenic mouse and rat models and characterized and compared digenic, monogenic, and wild-type phenotypes. Results The genetic interaction was synergistic in both species, with digenic animals exhibiting pheno- types of rapidly progressive PKD and early lethality resembling classic ARPKD. Genetic interaction be- tween Pkhd1 and Pkd1 depended on dosage in the digenic murine models, with no significant enhancement of the monogenic phenotype until a threshold of reduced expression at the second locus was breached. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

A CRISPR-Based Screen for Hedgehog Signaling Provides Insights Into Ciliary Function and Ciliopathies
ARTICLES https://doi.org/10.1038/s41588-018-0054-7 A CRISPR-based screen for Hedgehog signaling provides insights into ciliary function and ciliopathies David K. Breslow 1,2,7*, Sascha Hoogendoorn 3,7, Adam R. Kopp2, David W. Morgens4, Brandon K. Vu2, Margaret C. Kennedy1, Kyuho Han4, Amy Li4, Gaelen T. Hess4, Michael C. Bassik4, James K. Chen 3,5* and Maxence V. Nachury 2,6* Primary cilia organize Hedgehog signaling and shape embryonic development, and their dysregulation is the unifying cause of ciliopathies. We conducted a functional genomic screen for Hedgehog signaling by engineering antibiotic-based selection of Hedgehog-responsive cells and applying genome-wide CRISPR-mediated gene disruption. The screen can robustly identify factors required for ciliary signaling with few false positives or false negatives. Characterization of hit genes uncovered novel components of several ciliary structures, including a protein complex that contains δ -tubulin and ε -tubulin and is required for centriole maintenance. The screen also provides an unbiased tool for classifying ciliopathies and showed that many congenital heart disorders are caused by loss of ciliary signaling. Collectively, our study enables a systematic analysis of ciliary function and of ciliopathies, and also defines a versatile platform for dissecting signaling pathways through CRISPR-based screening. he primary cilium is a surface-exposed microtubule-based approach. Indeed, most studies to date have searched for genes that compartment that serves as an organizing center for diverse either intrinsically affect cell growth or affect sensitivity to applied Tsignaling pathways1–3. Mutations affecting cilia cause ciliopa- perturbations16–23. thies, a group of developmental disorders including Joubert syn- Here, we engineered a Hh-pathway-sensitive reporter to enable drome, Meckel syndrome (MKS), nephronophthisis (NPHP), and an antibiotic-based selection platform. -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -
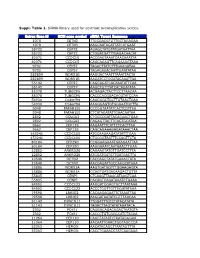
Suppl. Table 1
Suppl. Table 1. SiRNA library used for centriole overduplication screen. Entrez Gene Id NCBI gene symbol siRNA Target Sequence 1070 CETN3 TTGCGACGTGTTGCTAGAGAA 1070 CETN3 AAGCAATAGATTATCATGAAT 55722 CEP72 AGAGCTATGTATGATAATTAA 55722 CEP72 CTGGATGATTTGAGACAACAT 80071 CCDC15 ACCGAGTAAATCAACAAATTA 80071 CCDC15 CAGCAGAGTTCAGAAAGTAAA 9702 CEP57 TAGACTTATCTTTGAAGATAA 9702 CEP57 TAGAGAAACAATTGAATATAA 282809 WDR51B AAGGACTAATTTAAATTACTA 282809 WDR51B AAGATCCTGGATACAAATTAA 55142 CEP27 CAGCAGATCACAAATATTCAA 55142 CEP27 AAGCTGTTTATCACAGATATA 85378 TUBGCP6 ACGAGACTACTTCCTTAACAA 85378 TUBGCP6 CACCCACGGACACGTATCCAA 54930 C14orf94 CAGCGGCTGCTTGTAACTGAA 54930 C14orf94 AAGGGAGTGTGGAAATGCTTA 5048 PAFAH1B1 CCCGGTAATATCACTCGTTAA 5048 PAFAH1B1 CTCATAGATATTGAACAATAA 2802 GOLGA3 CTGGCCGATTACAGAACTGAA 2802 GOLGA3 CAGAGTTACTTCAGTGCATAA 9662 CEP135 AAGAATTTCATTCTCACTTAA 9662 CEP135 CAGCAGAAAGAGATAAACTAA 153241 CCDC100 ATGCAAGAAGATATATTTGAA 153241 CCDC100 CTGCGGTAATTTCCAGTTCTA 80184 CEP290 CCGGAAGAAATGAAGAATTAA 80184 CEP290 AAGGAAATCAATAAACTTGAA 22852 ANKRD26 CAGAAGTATGTTGATCCTTTA 22852 ANKRD26 ATGGATGATGTTGATGACTTA 10540 DCTN2 CACCAGCTATATGAAACTATA 10540 DCTN2 AACGAGATTGCCAAGCATAAA 25886 WDR51A AAGTGATGGTTTGGAAGAGTA 25886 WDR51A CCAGTGATGACAAGACTGTTA 55835 CENPJ CTCAAGTTAAACATAAGTCAA 55835 CENPJ CACAGTCAGATAAATCTGAAA 84902 CCDC123 AAGGATGGAGTGCTTAATAAA 84902 CCDC123 ACCCTGGTTGTTGGATATAAA 79598 LRRIQ2 CACAAGAGAATTCTAAATTAA 79598 LRRIQ2 AAGGATAATATCGTTTAACAA 51143 DYNC1LI1 TTGGATTTGTCTATACATATA 51143 DYNC1LI1 TAGACTTAGTATATAAATACA 2302 FOXJ1 CAGGACAGACAGACTAATGTA -

Novel Gene Discovery in Primary Ciliary Dyskinesia
Novel Gene Discovery in Primary Ciliary Dyskinesia Mahmoud Raafat Fassad Genetics and Genomic Medicine Programme Great Ormond Street Institute of Child Health University College London A thesis submitted in conformity with the requirements for the degree of Doctor of Philosophy University College London 1 Declaration I, Mahmoud Raafat Fassad, confirm that the work presented in this thesis is my own. Where information has been derived from other sources, I confirm that this has been indicated in the thesis. 2 Abstract Primary Ciliary Dyskinesia (PCD) is one of the ‘ciliopathies’, genetic disorders affecting either cilia structure or function. PCD is a rare recessive disease caused by defective motile cilia. Affected individuals manifest with neonatal respiratory distress, chronic wet cough, upper respiratory tract problems, progressive lung disease resulting in bronchiectasis, laterality problems including heart defects and adult infertility. Early diagnosis and management are essential for better respiratory disease prognosis. PCD is a highly genetically heterogeneous disorder with causal mutations identified in 36 genes that account for the disease in about 70% of PCD cases, suggesting that additional genes remain to be discovered. Targeted next generation sequencing was used for genetic screening of a cohort of patients with confirmed or suggestive PCD diagnosis. The use of multi-gene panel sequencing yielded a high diagnostic output (> 70%) with mutations identified in known PCD genes. Over half of these mutations were novel alleles, expanding the mutation spectrum in PCD genes. The inclusion of patients from various ethnic backgrounds revealed a striking impact of ethnicity on the composition of disease alleles uncovering a significant genetic stratification of PCD in different populations. -

Whole Exome Sequencing Reveals NOTCH1 Mutations in Anaplastic Large Cell Lymphoma and Points to Notch Both As a Key Pathway and a Potential Therapeutic Target
Non-Hodgkin Lymphoma SUPPLEMENTARY APPENDIX Whole exome sequencing reveals NOTCH1 mutations in anaplastic large cell lymphoma and points to Notch both as a key pathway and a potential therapeutic target Hugo Larose, 1,2 Nina Prokoph, 1,2 Jamie D. Matthews, 1 Michaela Schlederer, 3 Sandra Högler, 4 Ali F. Alsulami, 5 Stephen P. Ducray, 1,2 Edem Nuglozeh, 6 Mohammad Feroze Fazaludeen, 7 Ahmed Elmouna, 6 Monica Ceccon, 2,8 Luca Mologni, 2,8 Carlo Gambacorti-Passerini, 2,8 Gerald Hoefler, 9 Cosimo Lobello, 2,10 Sarka Pospisilova, 2,10,11 Andrea Janikova, 2,11 Wilhelm Woessmann, 2,12 Christine Damm-Welk, 2,12 Martin Zimmermann, 13 Alina Fedorova, 14 Andrea Malone, 15 Owen Smith, 15 Mariusz Wasik, 2,16 Giorgio Inghirami, 17 Laurence Lamant, 18 Tom L. Blundell, 5 Wolfram Klapper, 19 Olaf Merkel, 2,3 G. A. Amos Burke, 20 Shahid Mian, 6 Ibraheem Ashankyty, 21 Lukas Kenner 2,3,22 and Suzanne D. Turner 1,2,10 1Department of Pathology, University of Cambridge, Cambridge, UK; 2European Research Initiative for ALK Related Malignancies (ERIA; www.ERIALCL.net ); 3Department of Pathology, Medical University of Vienna, Vienna, Austria; 4Unit of Laboratory Animal Pathology, Uni - versity of Veterinary Medicine Vienna, Vienna, Austria; 5Department of Biochemistry, University of Cambridge, Tennis Court Road, Cam - bridge, UK; 6Molecular Diagnostics and Personalised Therapeutics Unit, Colleges of Medicine and Applied Medical Sciences, University of Ha’il, Ha’il, Saudi Arabia; 7Neuroinflammation Research Group, Department of Neurobiology, A.I Virtanen Institute -

Investigating Mammalian Development Using Mouse Forward Genetics
INVESTIGATING MAMMALIAN DEVELOPMENT USING MOUSE FORWARD GENETICS A Dissertation Presented to the Faculty of the Graduate School of Cornell University in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy by Joel Michael Brown May 2018 © 2018 Joel Michael Brown INVESTIGATING MAMMALIAN DEVELOPMENT USING MOUSE FORWARD GENETICS Joel Michael Brown, Ph.D. Cornell University 2018 Forward genetics allows the identification of novel genes involved in a particular biological process. We performed an ENU forward mutagenesis screen in mice aimed at identifying genes which regulate early organ morphogenesis during development. Fifteen mouse mutants were identified in the screen which possess cardiovascular, craniofacial, extraembryonic, or general body morphology defects. After performing preliminary characterization of all 15 lines, we identified a likely causative mutation in 14 of the 15 mutants. 1D and 13B mutants both exhibit craniofacial defects and were selected for in-depth characterization to identify the molecular mechanisms whereby they regulate embryogenesis. 1D mutants are characterized by an open neural tube in the hindbrain region. This phenotype is similar to human embryos with exencephaly, a congenital birth malformation of high incidence in the human population. Positional cloning of 1D identified a mutation in SPCA1, a Golgi-localized pump that controls calcium homeostasis. Results from the molecular characterization of mouse 1D mutants, as well as from time lapse microscopy of chicken embryos, revealed that calcium is tightly regulated during neural tube closure and that calcium homeostasis is required to promote apical constriction of neuroepithelial cells. These results show that SPCA1 activity is required to regulate the actomyosin dynamics that propel apical constriction, and that the actin severing protein, Cofilin 1, is a key mediator of SPCA1 function. -

Identification of Genomic Targets of Krüppel-Like Factor 9 in Mouse Hippocampal
Identification of Genomic Targets of Krüppel-like Factor 9 in Mouse Hippocampal Neurons: Evidence for a role in modulating peripheral circadian clocks by Joseph R. Knoedler A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy (Neuroscience) in the University of Michigan 2016 Doctoral Committee: Professor Robert J. Denver, Chair Professor Daniel Goldman Professor Diane Robins Professor Audrey Seasholtz Associate Professor Bing Ye ©Joseph R. Knoedler All Rights Reserved 2016 To my parents, who never once questioned my decision to become the other kind of doctor, And to Lucy, who has pushed me to be a better person from day one. ii Acknowledgements I have a huge number of people to thank for having made it to this point, so in no particular order: -I would like to thank my adviser, Dr. Robert J. Denver, for his guidance, encouragement, and patience over the last seven years; his mentorship has been indispensable for my growth as a scientist -I would also like to thank my committee members, Drs. Audrey Seasholtz, Dan Goldman, Diane Robins and Bing Ye, for their constructive feedback and their willingness to meet in a frequently cold, windowless room across campus from where they work -I am hugely indebted to Pia Bagamasbad and Yasuhiro Kyono for teaching me almost everything I know about molecular biology and bioinformatics, and to Arasakumar Subramani for his tireless work during the home stretch to my dissertation -I am grateful for the Neuroscience Program leadership and staff, in particular -

Identification of Two Novel Pathogenic Variants of PIBF1 by Whole Exome
Shen et al. BMC Medical Genetics (2020) 21:192 https://doi.org/10.1186/s12881-020-01130-x CASE REPORT Open Access Identification of two novel pathogenic variants of PIBF1 by whole exome sequencing in a 2-year-old boy with Joubert syndrome Yue Shen1,2†, Hao Wang3†, Zhimin Liu4†, Minna Luo1,2, Siyu Ma1,2, Chao Lu1,2, Zongfu Cao1,2, Yufei Yu1,2, Ruikun Cai1,2, Cuixia Chen1,2, Qian Li1,2, Huafang Gao1,2, Yun Peng4, Baoping Xu3* and Xu Ma1,2* Abstract Background: Joubert syndrome (OMIM 213300) is an autosomal recessive disorder with gene heterogeneity. Causal genes and their variants have been identified by sequencing or other technologies for Joubert syndrome subtypes. Case presentation: A two-year-old boy was diagnosed with Joubert syndrome by global development delay and molar tooth sign of mid-brain. Whole exome sequencing was performed to detect the causative gene variants in this individual, and the candidate pathogenic variants were verified by Sanger sequencing. We identified two pathogenic variants (NM_006346.2: c.1147delC and c.1054A > G) of PIBF1 in this Joubert syndrome individual, which is consistent with the mode of autosomal recessive inheritance. Conclusion: In this study, we identified two novel pathogenic variants in PIBF1 in a Joubert syndrome individual using whole exome sequencing, thereby expanding the PIBF1 pathogenic variant spectrum of Joubert syndrome. Keywords: PIBF1, Joubert syndrome, Cerebellar vermis hypoplasia, Whole exome sequencing Background many of the clinical features of ciliopathies [3], and Joubert syndrome (OMIM: 213300) is an autosomal many ciliary/basal body genes and variants have been recessive disorder characterized by a specific mid- discovered to be associated with Joubert syndrome [4, hindbrain malformation, hypotonia and developmental 5]. -

Chromatin Conformation Links Distal Target Genes to CKD Loci
BASIC RESEARCH www.jasn.org Chromatin Conformation Links Distal Target Genes to CKD Loci Maarten M. Brandt,1 Claartje A. Meddens,2,3 Laura Louzao-Martinez,4 Noortje A.M. van den Dungen,5,6 Nico R. Lansu,2,3,6 Edward E.S. Nieuwenhuis,2 Dirk J. Duncker,1 Marianne C. Verhaar,4 Jaap A. Joles,4 Michal Mokry,2,3,6 and Caroline Cheng1,4 1Experimental Cardiology, Department of Cardiology, Thoraxcenter Erasmus University Medical Center, Rotterdam, The Netherlands; and 2Department of Pediatrics, Wilhelmina Children’s Hospital, 3Regenerative Medicine Center Utrecht, Department of Pediatrics, 4Department of Nephrology and Hypertension, Division of Internal Medicine and Dermatology, 5Department of Cardiology, Division Heart and Lungs, and 6Epigenomics Facility, Department of Cardiology, University Medical Center Utrecht, Utrecht, The Netherlands ABSTRACT Genome-wide association studies (GWASs) have identified many genetic risk factors for CKD. However, linking common variants to genes that are causal for CKD etiology remains challenging. By adapting self-transcribing active regulatory region sequencing, we evaluated the effect of genetic variation on DNA regulatory elements (DREs). Variants in linkage with the CKD-associated single-nucleotide polymorphism rs11959928 were shown to affect DRE function, illustrating that genes regulated by DREs colocalizing with CKD-associated variation can be dysregulated and therefore, considered as CKD candidate genes. To identify target genes of these DREs, we used circular chro- mosome conformation capture (4C) sequencing on glomerular endothelial cells and renal tubular epithelial cells. Our 4C analyses revealed interactions of CKD-associated susceptibility regions with the transcriptional start sites of 304 target genes. Overlap with multiple databases confirmed that many of these target genes are involved in kidney homeostasis.