Highly Variable Chloroplast Genome from Two Endangered Papaveraceae Lithophytes Corydalis Saxicola and C
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
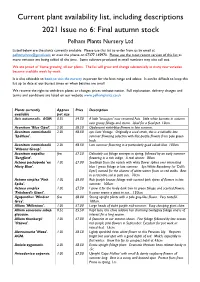
Current Plant Availability List, Including Descriptions 2021 Issue No 6: Final Autumn Stock Pelham Plants Nursery Ltd
Current plant availability list, including descriptions 2021 Issue no 6: Final autumn stock Pelham Plants Nursery Ltd Listed below are the plants currently available. Please use this list to order from us by email at [email protected] or over the phone on 07377 145970. Please use the most recent version of this list as more varieties are being added all the time. Some cultivars produced in small numbers may also sell out. We are proud of ‘home growing’ all our plants. The list will grow and change substantially as many new varieties become available week by week. It is also advisable to book to visit the nursery in person for the best range and advice. It can be difficult to keep this list up to date at our busiest times or when batches are small. We reserve the right to withdraw plants or changes prices without notice. Full explanation, delivery charges and terms and conditions are listed on our website www.pelhamplants.co.uk Plants currently Approx Price Description available pot size Acis autumnalis. AGM. 0.5L £4.50 A little 'Leucojum' now renamed Acis. Little white bonnets in autumn over grassy foliage and stems. Ideal for a focal pot. 10cm. Aconitum 'Blue Opal'. 2.0L £8.50 Opalescent violet-blue flowers in late summer. Aconitum carmichaelii 2.0L £8.50 syn. Late Vintage. Originally a seed strain, this is a valuable late 'Spätlese'. summer flowering selection with lilac-purple flowers from pale green buds. Aconitum carmichaelii 2.0L £8.50 Late summer flowering in a particularly good cobalt blue. -

Isolation and Identification of Alkaloids from Macleaya Microcarpa by UHPLC–Q-TOF-MS and Their Cytotoxic Activity in Vitro, An
Sai et al. BMC Chemistry (2020) 14:5 https://doi.org/10.1186/s13065-020-0660-1 BMC Chemistry RESEARCH ARTICLE Open Access Isolation and identifcation of alkaloids from Macleaya microcarpa by UHPLC– Q-TOF-MS and their cytotoxic activity in vitro, antiangiogenic activity in vivo Chunmei Sai1,2, Jian’an Wang1, Binjie Li3, Lin Ding1, Huiyun Wang1, Qibao Wang1, Huiming Hua3, Fangpeng Zhang2 and Qiang Ren1* Abstract Background: Extensive bioactivities of alkaloids from the genus Macleaya (Macleaya cordata (Willd.) R. Br. and Macleaya microcarpa (Maxim.) Fedde) have been widely reported, as well as more and more concerned from the sci- entifc communities. However, systematic research on the phytochemical information of M. microcarpa is incomplete. The aim of this study was to rapidly and conveniently qualitative analyze alkaloids from M. microcarpa by ultra-perfor- mance liquid chromatography/quadrupole-time-of-fght mass spectrometry (UHPLC–Q-TOF-MS) using accurate mass weight and characteristic fragment ions, furthermore separate and identify the main alkaloids, test antitumor activity in vitro and antiangiogenic activity in vivo. Results: A total of 14 alkaloids from fruits of M. microcarpa were identifed by UHPLC–Q-TOF-MS, including 5 pro- topines, 2 benzophenanthridines, 1 dimer, 1 dihydrobenzophenanthridines and 5 unknown structure compounds. Two major alkaloids were isolated by various column chromatographic methods. Their structures were determined by NMR data and related literatures. The two major alkaloids were evaluated for intro cytotoxic activities against HL-60, MCF-7, A-549, and in vivo antiangiogenic activity using transgenic zebrafsh. Conclusions: Current qualitative method based on UHPLC–Q-TOF-MS technique provided a scientifc basis for isola- tion, structural identifcation, and in vitro or in vivo pharmacological further study of alkaloids from M. -

Berberine: Botanical Occurrence, Traditional Uses, Extraction Methods, and Relevance in Cardiovascular, Metabolic, Hepatic, and Renal Disorders
REVIEW published: 21 August 2018 doi: 10.3389/fphar.2018.00557 Berberine: Botanical Occurrence, Traditional Uses, Extraction Methods, and Relevance in Cardiovascular, Metabolic, Hepatic, and Renal Disorders Maria A. Neag 1, Andrei Mocan 2*, Javier Echeverría 3, Raluca M. Pop 1, Corina I. Bocsan 1, Gianina Cri¸san 2 and Anca D. Buzoianu 1 1 Department of Pharmacology, Toxicology and Clinical Pharmacology, “Iuliu Hatieganu” University of Medicine and Pharmacy, Cluj-Napoca, Romania, 2 Department of Pharmaceutical Botany, “Iuliu Hatieganu” University of Medicine and Pharmacy, Cluj-Napoca, Romania, 3 Department of Environmental Sciences, Universidad de Santiago de Chile, Santiago de Chile, Chile Edited by: Berberine-containing plants have been traditionally used in different parts of the world for Anna Karolina Kiss, the treatment of inflammatory disorders, skin diseases, wound healing, reducing fevers, Medical University of Warsaw, Poland affections of eyes, treatment of tumors, digestive and respiratory diseases, and microbial Reviewed by: Pinarosa Avato, pathologies. The physico-chemical properties of berberine contribute to the high diversity Università degli Studi di Bari Aldo of extraction and detection methods. Considering its particularities this review describes Moro, Italy various methods mentioned in the literature so far with reference to the most important Sylwia Zielinska, Wroclaw Medical University, Poland factors influencing berberine extraction. Further, the common separation and detection *Correspondence: methods like thin layer chromatography, high performance liquid chromatography, and Andrei Mocan mass spectrometry are discussed in order to give a complex overview of the existing [email protected] methods. Additionally, many clinical and experimental studies suggest that berberine Specialty section: has several pharmacological properties, such as immunomodulatory, antioxidative, This article was submitted to cardioprotective, hepatoprotective, and renoprotective effects. -

Bocconia and Macleaya Author(S): J
Bocconia and Macleaya Author(s): J. Hutchinson Source: Bulletin of Miscellaneous Information (Royal Botanic Gardens, Kew), Vol. 1920, No. 8 (1920), pp. 275-282 Published by: Springer on behalf of Royal Botanic Gardens, Kew Stable URL: http://www.jstor.org/stable/4120232 Accessed: 14-06-2016 14:47 UTC Your use of the JSTOR archive indicates your acceptance of the Terms & Conditions of Use, available at http://about.jstor.org/terms JSTOR is a not-for-profit service that helps scholars, researchers, and students discover, use, and build upon a wide range of content in a trusted digital archive. We use information technology and tools to increase productivity and facilitate new forms of scholarship. For more information about JSTOR, please contact [email protected]. Royal Botanic Gardens, Kew, Springer are collaborating with JSTOR to digitize, preserve and extend access to Bulletin of Miscellaneous Information (Royal Botanic Gardens, Kew) This content downloaded from 131.172.36.29 on Tue, 14 Jun 2016 14:47:17 UTC All use subject to http://about.jstor.org/terms 275 an old but no longer common species, 28 ft. high and 5 ft. in girth of trunk; and a fine healthy tree of the newer Japanese M. hypoleuca over 30 ft. high. There is a good collection of hardy bamboos on the lower, damper part of the garden, and it was interesting to see Phyllo- stachys aurea flowering, although not gratifying, since it portends the flowering and consequent death of the species throughout the country. XLIV.-BOCCONIA AND MACLEAYA. J. HUTCHINSON. The genus Bocconia (Papaveraceae) was founded by Linnaeus* in 1737, the type species being B. -

Identification of the Impurities in Bopu Powder® and Sangrovit® by LC
molecules Article Identification of the Impurities in Bopu Powder® and Sangrovit® by LC-MS Combined with a Screening Method Zhuang Dong 1,2,†, Mengting Liu 1,3,†, Xiaohong Zhong 2, Xiaoyong Ou 1,3, Xuan Yun 1,3, Mingcan Wang 1, Shurui Ren 1, Zhixing Qing 1,3,* and Jianguo Zeng 1,3,* 1 Hunan Key Laboratory of Traditional Chinese Veterinary Medicine, Hunan Agricultural University, Changsha 410128, China; [email protected] (Z.D.); [email protected] (M.L.); [email protected] (X.O.); [email protected] (X.Y.); [email protected] (M.W.); [email protected] (S.R.) 2 College of Horticulture, Hunan Agricultural University, Changsha 410128, China; [email protected] 3 College of Veterinary Medicine, Hunan Agricultural University, Changsha 410128, China * Correspondence: [email protected] (Z.Q.); [email protected] (J.Z.); Tel./Fax: +86-0731-84686560 (Z.Q. & J.Z.) † These authors contributed equally to this work. Abstract: Bopu powder® and Sangrovit® were developed from Macleaya cordata and are widely used in agriculture and animal husbandry, but their impurities have been rarely reported in the literature. Impurity analysis is of great importance to the quality and safety of veterinary drugs. In this study, high-performance liquid chromatography/quadrupole time-of-flight mass spectrometry (HPLC-Q-TOF-MS) combined with a screening method was used to screen and characterize the impurities in Bopu powder® and Sangrovit®. A total of 58 impurities were screened from Bopu Citation: Dong, Z.; Liu, M.; Zhong, powder® and Sangrovit® using the screening strategies, of which 39 were identified by their accurate X.; Ou, X.; Yun, X.; Wang, M.; Ren, S.; m/z value, characteristic MS/MS data, and fragmentation pathways of references. -

Evolution of Reproductive Morphology in the Papaveraceae S.L. (Papaveraceae and Fumariaceae, Ranunculales)
® International Journal of Plant Developmental Biology ©2010 Global Science Books Evolution of Reproductive Morphology in the Papaveraceae s.l. (Papaveraceae and Fumariaceae, Ranunculales) Oriane Hidalgo • Stefan Gleissberg* Department of Environmental and Plant Biology, Ohio University, 500 Porter Hall, Athens, Ohio 45701, USA Corresponding author : * [email protected] ABSTRACT Flower bearing branching systems are of major importance for plant reproduction, and exhibit significant variation between and within lineages. A key goal in evolutionary biology is to discover and characterize changes in the genetic programming of development that drive the modification and diversification of morphology. Here we present a synopsis of reproductive architecture in Papaveraceae s.l., a lineage in which the evolution of inflorescence determinacy, flower structure and symmetry, and effloration sequence produced unique reproduc- tive syndromes. We discuss the potential of this group to study key issues on the evolution of reproductive structures, and refer to candidate gene families, choice of landmark species, and available tools for developmental genetic investigations. _____________________________________________________________________________________________________________ Keywords: effloration sequence, flower organ identity, flower symmetry, Fumariaceae, inflorescence determinacy Abbreviations: AP1, APETALA1; AP3, APETALA3; CEN, CENTRORADIALIS; CRC, CRABS CLAW; CYC, CYCLOIDEA; FLO, FLORICAULA; FUL, FRUITFULL; LFY, LEAFY; PI, PISTILLATA; TFL1, -

The Production of Isoquinoline Alkaloids by Plant Sell
Vi* · <P620 7-SS2 The Chemistry and Biology of Isoquinoline Alkaloids Edited by J. D. Phillipson, M.F. Roberts, and Μ. H. Zenk With 178 Figures Springer-Verlag Berlin Heidelberg NewYork Tokyo Contents Plants as a Source of Isoquinoline Alkaloids N.G. Bisset (With 5 Figures) 1 Chemotaxonomy of the Papaveraceae Alkaloids V. Preininger (With 8 Figures) 23 Structure Activities and Pharmacological Properties of the Opium Alkaloids E. Lindner (With 7 Figures) 38 The Occurrence of Simple Isoquinolines in Plants J. Lundström (With 3 Figures) 47 Erythnna Alkaloids AÜ. Jackson (With 12 Figures) 62 Annonaceae Alkaloids A. Cave (With 22 Figures) 79 The Chemistry and Pharmacology of Cularine Alkaloids L. Castedo (With 28 Figures) 102 Bisbenzylisoquinoline Alkaloids P.L. Schiff, Jr. (With 5 Figures) 126 Natural Degradative Routes for the Aporphines M. Shamma (With 5 Figures) 142 Synthesis and Structure-Activity Relationships of Apoq)hines as Dopamine Receptor Agonists and Antagonists J.L. Neumeyer (With 8 Figures) 146 The Chemistry and Pharmacology of Morphinan Alkaloids A. Brossi (With 15 Figures) 171 VIII Contents The Development of a Practical Total Synthesis of Natural and Unnatural Codeine, Morphine and Thebaine K.C.Rice (With 9Figures) 191 Biomimetic and Total Synthesis of Monoterpenoid Isoquinoline Alkaloids R.T. Brown (With 7 Figures) 204 The Biosynthesis of Isoquinoline Alkaloids R.B. Herbert (With 14 Figures) 213 Biosynthesis of Morphinan Alkaloids E. Brochmann-Hanssen (With 7 Figures) 229 Enzymology of Benzylisoquinoline Alkaloid Formation MB. Zenk (With 12 Figures) 240 Morphinan Alkaloids from Plant Cell Cultures F. Constabel 257 The Production of Isoquinoline Alkaloids by Plant Cell Cultures M. -

Sanguinaria Canadensis: Traditional Medicine, Phytochemical Composition, Biological Activities and Current Uses
International Journal of Molecular Sciences Review Sanguinaria canadensis: Traditional Medicine, Phytochemical Composition, Biological Activities and Current Uses Andrew Croaker 1,*, Graham J. King 1, John H. Pyne 2, Shailendra Anoopkumar-Dukie 3 and Lei Liu 1,* 1 Southern Cross Plant Science, Southern Cross University, Lismore, NSW 2480, Australia; [email protected] 2 School of Medicine, The University of Queensland, St. Lucia, QLD 4072, Australia; [email protected] 3 School of Pharmacy, Griffith University, Gold Coast Campus, Gold Coast, QLD 4222, Australia; s.dukie@griffith.edu.au * Correspondence: [email protected] (A.C.); [email protected] (L.L.); Tel.: +61-04-2959-4932 (A.C.); +61-02-6622-3211 (L.L.) Academic Editor: Chang Won Choi Received: 22 July 2016; Accepted: 12 August 2016; Published: 27 August 2016 Abstract: Sanguinaria canadensis, also known as bloodroot, is a traditional medicine used by Native Americans to treat a diverse range of clinical conditions. The plants rhizome contains several alkaloids that individually target multiple molecular processes. These bioactive compounds, mechanistically correlate with the plant’s history of ethnobotanical use. Despite their identification over 50 years ago, the alkaloids of S. canadensis have not been developed into successful therapeutic agents. Instead, they have been associated with clinical toxicities ranging from mouthwash induced leukoplakia to cancer salve necrosis and treatment failure. This review explores the historical use of S. canadensis, the molecular actions of the benzophenanthridine and protopin alkaloids it contains, and explores natural alkaloid variation as a possible rationale for the inconsistent efficacy and toxicities encountered by S. -

Considérations Sur L'histoire Naturelle Des Ranunculales
Considérations sur l’histoire naturelle des Ranunculales Laetitia Carrive To cite this version: Laetitia Carrive. Considérations sur l’histoire naturelle des Ranunculales. Botanique. Université Paris-Saclay, 2019. Français. NNT : 2019SACLS177. tel-02276988 HAL Id: tel-02276988 https://tel.archives-ouvertes.fr/tel-02276988 Submitted on 3 Sep 2019 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Considérations sur l’histoire naturelle des Ranunculales 2019SACLS177 Thèse de doctorat de l'Université Paris-Saclay : préparée à l’Université Paris-Sud NNT École doctorale n°567 : Sciences du végétal, du gène à l'écosystème (SDV) Spécialité de doctorat : Biologie Thèse présentée et soutenue à Orsay, le 05 juillet 2019, par Laetitia Carrive Composition du Jury : Catherine Damerval Directrice de recherche, CNRS (– UMR 320 GQE) Présidente du jury Julien Bachelier Professeur, Freie Universität Berlin (– Institute of Biology) Rapporteur Thomas Haevermans Maître de conférences, MNHN (– UMR 7205 ISYEB) Rapporteur Jean-Yves Dubuisson Professeur, SU (–UMR 7205 ISYEB) Examinateur Sophie Nadot Professeure, U-PSud (– UMR 8079 ESE) Directrice de thèse « Le commencement sera d’admirer tout, même les choses les plus communes. Le milieu, d’écrire ce que l’on a bien vu et ce qui est d’utilité. -

Eastern Washington Garden Wise Non-Invasive Plants for Your Garden
Garden Wise Non-Invasive Plants for Your Garden Spring 2010 Eastern Washington Guide Voluntary codes of conduct 'PSUIF(BSEFOJOH1VCMJD BOOPUBUFE *OBOFêPSUUPSFEVDFUIFTQSFBEPGJOWBTJWFQMBOUTVTFEGPS IPSUJDVMUVSBMQVSQPTFT FYQFSUTIBWFDSFBUFEUIFi7PMVOUBSZ $PEFTPG$POEVDU wBTFSJFTPGTUFQTUIBUOVSTFSZQSPGFTTJPOBMT MBOETDBQFBSDIJUFDUT HBSEFOFST BOEPUIFSTDBOUBLFUPIFMQDVSC UIFTQSFBEPGJOWBTJWFIPSUJDVMUVSBMQMBOUT Ǚ"TLGPSPOMZOPOJOWBTJWFTQFDJFTXIFOZPVBDRVJSFQMBOUT 1MBOUPOMZFOWJSPONFOUBMMZTBGFTQFDJFTJOZPVSHBSEFOT 8PSLUPXBSETBOEQSPNPUFOFXMBOETDBQFEFTJHOUIBUJT GSJFOEMZUPSFHJPOBMFDPTZTUFNT Ǚ4FFLJOGPSNBUJPOPOXIJDITQFDJFTBSFJOWBTJWFJOZPVSBSFB 4PVSDFTDPVMEJODMVEFCPUBOJDBMHBSEFOT IPSUJDVMUVSJTUT DPOTFSWBUJPOJTUT BOEHPWFSONFOUBHFODJFT3FNPWF JOWBTJWFTQFDJFTGSPNZPVSMBOEBOESFQMBDFUIFNXJUIOPO JOWBTJWFTQFDJFTTVJUFEUPZPVSTJUFBOEOFFET Ǚ%POPUUSBEFQMBOUTXJUIPUIFSHBSEFOFSTJGZPVLOPXUIFZ BSFTQFDJFTXJUIJOWBTJWFDIBSBDUFSJTUJDT Ǚ3FRVFTUUIBUCPUBOJDBMHBSEFOTBOEOVSTFSJFTQSPNPUF EJTQMBZ BOETFMMPOMZOPOJOWBTJWFTQFDJFT Ǚ)FMQFEVDBUFZPVSDPNNVOJUZBOEPUIFSHBSEFOFSTJO ZPVSBSFBUISPVHIQFSTPOBMDPOUBDUBOEJOTVDITFUUJOHTBT HBSEFODMVCTBOEPUIFSDJWJDHSPVQT 'PSUIFGVMM(BSEFOJOH$PEFTPG$POEVDU PSUPMFBSOBCPVU UIF$PEFTPG$POEVDUGPS(PWFSONFOU /VSTFSZ1SPGFTTJPOBMT -BOETDBQF"SDIJUFDUT BOE#PUBOJD(BSEFOTBOE"SCPSFUB QMFBTF HPUPUIF$FOUFSGPS1MBOU$POTFSWBUJPOTXFCTJUFBU XXXDFOUFSGPSQMBOUDPOTFSWBUJPOPSHJOWBTJWFTDPEFTOIUNM Garden Wise is dedicated to Ann Lennartz Garden Wise Non-Invasive Plants for Your Garden 8IJMFNPTUFYPUJDQMBOUTBSFOPUQSPCMFNBUJD BGFXIBWF CFDPNFJOWBTJWFJO8BTIJOHUPO4UBUF8IFOUIFTFQMBOUT TQSFBEUPXJMEBOEBHSJDVMUVSBMBSFBT -

2012Catalogfinal.Pdf
Phone (603) 463-7663 Fax (603) 463-7326 www.vanberkumnursery.com email: [email protected] At Van Berkum Nursery, we strive for continued growth as a source for healthy perennials in New England, to stay abreast of the latest developments in our field, and to retain a family-run business of integrity. Greetings, We are environmentally friendly. Check out the details are environmentally friendly. We in the “Why Van Berkum Nursery” in the “Why Van subpage on our What a year! A busy April, rainy May, decent summer and a wild fall. We thank you for your business this past year. This may be a no-brainer, but it is because of you that we staying ahead in these tough times. We are doing a two-year catalog for 2012-13 since many of you are using the computer to access our information; we can also save in paper, costs, and time. We have taken the prices out of the text (see price sheet inside the back cover) so you website. can share the catalog with your clients. Next year, we’ll have a supplement for 2013 with our new plants for the year. Your clients are also welcome to use our website to access perennial information; many people have commented on the printable plant list feature under photos that lets you create a pdf of selected plants. We’d love to see you on Facebook, and if you don’t receive our weekly e-mails during the season (bi-monthly in winter) and would like to, please sign up on our healthy and tough going into the winter. -

Two-Year Catalog WHOLESALE PERENNIALS & GROUNDCOVERS
Two-Y ear Catalog 2014 – 2015 WHOLESALE PERENNIALS & GROUNDCOVERS Phone (603) 463-7663 Fax (603) 463-7326 www.vanberkumnursery.com email: [email protected] At Van Berkum Nursery, we strive for continued growth as a source for healthy perennials in New England, to stay abreast of the latest developments in our field, and to retain a family-run business of integrity. Greetings, Here is our new 2 year catalog for 2014-15. Next fall, our 2015 supplement will insert into the flap at the back of this catalog. Many of you are accessing info online, but there are still plenty of Ôtruck officesÕ out there that want a book version in their front seat files. We hope this one is on top...HereÕs whatÕs new: 1) We have been doing more and more custom growing for special jobs. If you have a landscape job that requires large numbers, or even plants we donÕt normally grow, give us call; we can price them competitively, and have special staff members who can be your contact for these orders. 2) We did a lot of construction over the past year. We built a large staging building, where your orders are prepared for shipment. We rebuilt our front office, combining 4 tiny rooms into one roomy one. And lastly, weÕve nearly completed our water recycling project; 90 % of our runoff is now reused in irrigation. 3) WeÕve introduced many new plants in the last couple of years and would love your feedback. What is working for you? Are there plants that youÕve used that you feel we should be growing? We learn so much from all of you who are Ôout there in the trenchesÕ so to speak.