Identification of Chemical Probes from Macleaya Cordata (Willd.) R.Br
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Developmental and Genetic Bases of Apetaly in Bocconia Frutescens
Arango‑Ocampo et al. EvoDevo (2016) 7:16 DOI 10.1186/s13227-016-0054-6 EvoDevo RESEARCH Open Access The developmental and genetic bases of apetaly in Bocconia frutescens (Chelidonieae: Papaveraceae) Cristina Arango‑Ocampo1, Favio González2, Juan Fernando Alzate3 and Natalia Pabón‑Mora1* Abstract Background: Bocconia and Macleaya are the only genera of the poppy family (Papaveraceae) lacking petals; how‑ ever, the developmental and genetic processes underlying such evolutionary shift have not yet been studied. Results: We studied floral development in two species of petal-less poppies Bocconia frutescens and Macleaya cordata as well as in the closely related petal-bearing Stylophorum diphyllum. We generated a floral transcriptome of B. frutescens to identify MADS-box ABCE floral organ identity genes expressed during early floral development. We performed phylogenetic analyses of these genes across Ranunculales as well as RT-PCR and qRT-PCR to assess loci- specific expression patterns. We found that petal-to-stamen homeosis in petal-less poppies occurs through distinct developmental pathways. Transcriptomic analyses of B. frutescens floral buds showed that homologs of all MADS-box genes are expressed except for the APETALA3-3 ortholog. Species-specific duplications of other ABCE genes inB. frute- scens have resulted in functional copies with expanded expression patterns than those predicted by the model. Conclusions: Petal loss in B. frutescens is likely associated with the lack of expression of AP3-3 and an expanded expression of AGAMOUS. The genetic basis of petal identity is conserved in Ranunculaceae and Papaveraceae although they have different number of AP3 paralogs and exhibit dissimilar floral groundplans. -
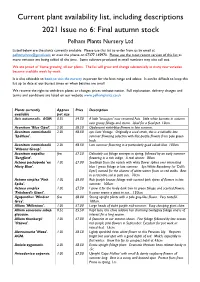
Current Plant Availability List, Including Descriptions 2021 Issue No 6: Final Autumn Stock Pelham Plants Nursery Ltd
Current plant availability list, including descriptions 2021 Issue no 6: Final autumn stock Pelham Plants Nursery Ltd Listed below are the plants currently available. Please use this list to order from us by email at [email protected] or over the phone on 07377 145970. Please use the most recent version of this list as more varieties are being added all the time. Some cultivars produced in small numbers may also sell out. We are proud of ‘home growing’ all our plants. The list will grow and change substantially as many new varieties become available week by week. It is also advisable to book to visit the nursery in person for the best range and advice. It can be difficult to keep this list up to date at our busiest times or when batches are small. We reserve the right to withdraw plants or changes prices without notice. Full explanation, delivery charges and terms and conditions are listed on our website www.pelhamplants.co.uk Plants currently Approx Price Description available pot size Acis autumnalis. AGM. 0.5L £4.50 A little 'Leucojum' now renamed Acis. Little white bonnets in autumn over grassy foliage and stems. Ideal for a focal pot. 10cm. Aconitum 'Blue Opal'. 2.0L £8.50 Opalescent violet-blue flowers in late summer. Aconitum carmichaelii 2.0L £8.50 syn. Late Vintage. Originally a seed strain, this is a valuable late 'Spätlese'. summer flowering selection with lilac-purple flowers from pale green buds. Aconitum carmichaelii 2.0L £8.50 Late summer flowering in a particularly good cobalt blue. -

Isolation and Identification of Alkaloids from Macleaya Microcarpa by UHPLC–Q-TOF-MS and Their Cytotoxic Activity in Vitro, An
Sai et al. BMC Chemistry (2020) 14:5 https://doi.org/10.1186/s13065-020-0660-1 BMC Chemistry RESEARCH ARTICLE Open Access Isolation and identifcation of alkaloids from Macleaya microcarpa by UHPLC– Q-TOF-MS and their cytotoxic activity in vitro, antiangiogenic activity in vivo Chunmei Sai1,2, Jian’an Wang1, Binjie Li3, Lin Ding1, Huiyun Wang1, Qibao Wang1, Huiming Hua3, Fangpeng Zhang2 and Qiang Ren1* Abstract Background: Extensive bioactivities of alkaloids from the genus Macleaya (Macleaya cordata (Willd.) R. Br. and Macleaya microcarpa (Maxim.) Fedde) have been widely reported, as well as more and more concerned from the sci- entifc communities. However, systematic research on the phytochemical information of M. microcarpa is incomplete. The aim of this study was to rapidly and conveniently qualitative analyze alkaloids from M. microcarpa by ultra-perfor- mance liquid chromatography/quadrupole-time-of-fght mass spectrometry (UHPLC–Q-TOF-MS) using accurate mass weight and characteristic fragment ions, furthermore separate and identify the main alkaloids, test antitumor activity in vitro and antiangiogenic activity in vivo. Results: A total of 14 alkaloids from fruits of M. microcarpa were identifed by UHPLC–Q-TOF-MS, including 5 pro- topines, 2 benzophenanthridines, 1 dimer, 1 dihydrobenzophenanthridines and 5 unknown structure compounds. Two major alkaloids were isolated by various column chromatographic methods. Their structures were determined by NMR data and related literatures. The two major alkaloids were evaluated for intro cytotoxic activities against HL-60, MCF-7, A-549, and in vivo antiangiogenic activity using transgenic zebrafsh. Conclusions: Current qualitative method based on UHPLC–Q-TOF-MS technique provided a scientifc basis for isola- tion, structural identifcation, and in vitro or in vivo pharmacological further study of alkaloids from M. -

Berberine: Botanical Occurrence, Traditional Uses, Extraction Methods, and Relevance in Cardiovascular, Metabolic, Hepatic, and Renal Disorders
REVIEW published: 21 August 2018 doi: 10.3389/fphar.2018.00557 Berberine: Botanical Occurrence, Traditional Uses, Extraction Methods, and Relevance in Cardiovascular, Metabolic, Hepatic, and Renal Disorders Maria A. Neag 1, Andrei Mocan 2*, Javier Echeverría 3, Raluca M. Pop 1, Corina I. Bocsan 1, Gianina Cri¸san 2 and Anca D. Buzoianu 1 1 Department of Pharmacology, Toxicology and Clinical Pharmacology, “Iuliu Hatieganu” University of Medicine and Pharmacy, Cluj-Napoca, Romania, 2 Department of Pharmaceutical Botany, “Iuliu Hatieganu” University of Medicine and Pharmacy, Cluj-Napoca, Romania, 3 Department of Environmental Sciences, Universidad de Santiago de Chile, Santiago de Chile, Chile Edited by: Berberine-containing plants have been traditionally used in different parts of the world for Anna Karolina Kiss, the treatment of inflammatory disorders, skin diseases, wound healing, reducing fevers, Medical University of Warsaw, Poland affections of eyes, treatment of tumors, digestive and respiratory diseases, and microbial Reviewed by: Pinarosa Avato, pathologies. The physico-chemical properties of berberine contribute to the high diversity Università degli Studi di Bari Aldo of extraction and detection methods. Considering its particularities this review describes Moro, Italy various methods mentioned in the literature so far with reference to the most important Sylwia Zielinska, Wroclaw Medical University, Poland factors influencing berberine extraction. Further, the common separation and detection *Correspondence: methods like thin layer chromatography, high performance liquid chromatography, and Andrei Mocan mass spectrometry are discussed in order to give a complex overview of the existing [email protected] methods. Additionally, many clinical and experimental studies suggest that berberine Specialty section: has several pharmacological properties, such as immunomodulatory, antioxidative, This article was submitted to cardioprotective, hepatoprotective, and renoprotective effects. -

Big Island Invasive Species Committee (BIISC) Highlights BIISC
Big Island Invasive Species Committee (BIISC) Highlights BIISC surveyed 6,244 acres for 5 key target species, and two incidental species and controlled a total of 16,608 acres, 17,610 individual plants were treated, and a total of 4,357 worker hours were used. An additional 165 miles of road were surveyed by the early detection crew looking for and mapping 138 potentially incipient invasive species. Miconia calvescens • BIISC continues to focus containment strategy along a 40-mile containment buffer between Malama Ki in lower Puna to Ninole in the Hamakua districts. Surveys focused in the Hilo, Hamakua and Puna districts. • Completed aerial surveys in the upper Hamakua district at elevations between 1,400' and 1,600'. A total of 1,598 acres were surveyed by air, while ground crews removed a total of 687 plants in this area. • Expanded survey and control effort from 1,600' to the 1,800' elevation above the core population in Onomea after finding a small number of plants at the 1,700' elevation. • Began work in a 52-acre control block in the Nanawale Forest Reserve. This project was terminated after data analysis revealed the feasibility to complete the mission was questionable after finding a large number of plants within the control area, as well as adjacent private parcels. • Completed survey and control blocks in the Makuu Forest Reserve. DOFAW donated 12 hours of air time to this effort which allowed BIISC to drop the ground crew at the far southern end of the survey sites to limit walk time to the survey and control blocks. -

Landscape Plants Rated by Deer Resistance
E271 Bulletin For a comprehensive list of our publications visit www.rce.rutgers.edu Landscape Plants Rated by Deer Resistance Pedro Perdomo, Morris County Agricultural Agent Peter Nitzsche, Morris County Agricultural Agent David Drake, Ph.D., Extension Specialist in Wildlife Management The following is a list of landscape plants rated according to their resistance to deer damage. The list was compiled with input from nursery and landscape professionals, Cooperative Extension personnel, and Master Gardeners in Northern N.J. Realizing that no plant is deer proof, plants in the Rarely Damaged, and Seldom Rarely Damaged categories would be best for landscapes prone to deer damage. Plants Occasionally Severely Damaged and Frequently Severely Damaged are often preferred by deer and should only be planted with additional protection such as the use of fencing, repellents, etc. Success of any of these plants in the landscape will depend on local deer populations and weather conditions. Latin Name Common Name Latin Name Common Name ANNUALS Petroselinum crispum Parsley Salvia Salvia Rarely Damaged Tagetes patula French Marigold Ageratum houstonianum Ageratum Tropaeolum majus Nasturtium Antirrhinum majus Snapdragon Verbena x hybrida Verbena Brugmansia sp. (Datura) Angel’s Trumpet Zinnia sp. Zinnia Calendula sp. Pot Marigold Catharanthus rosea Annual Vinca Occasionally Severely Damaged Centaurea cineraria Dusty Miller Begonia semperflorens Wax Begonia Cleome sp. Spider Flower Coleus sp. Coleus Consolida ambigua Larkspur Cosmos sp. Cosmos Euphorbia marginata Snow-on-the-Mountain Dahlia sp. Dahlia Helichrysum Strawflower Gerbera jamesonii Gerbera Daisy Heliotropium arborescens Heliotrope Helianthus sp. Sunflower Lobularia maritima Sweet Alyssum Impatiens balsamina Balsam, Touch-Me-Not Matricaria sp. False Camomile Impatiens walleriana Impatiens Myosotis sylvatica Forget-Me-Not Ipomea sp. -

Highly Variable Chloroplast Genome from Two Endangered Papaveraceae Lithophytes Corydalis Saxicola and C
Highly variable chloroplast genome from two endangered Papaveraceae lithophytes Corydalis saxicola and C. tomentella fengming Ren Chinese Academy of Medical Sciences & Peking Union Medical College Institute of Medicinal Plant Development Liqiang Wang Heze University Ying Li Chinese Academy of Medical Sciences & Peking Union Medical College Institute of Medicinal Plant Development Wei Zhuo Chongqing Insititute of Medicinal Plant Cultivation Zhichao Xu Chinese Academy of Medical Sciences & Peking Union Medical College Institute of Medicinal Plant Development Haojie Guo Wuhu Institute of Technology Yan Liu Chongqing Institute of Medicinal Plant Cultivation Ranran Gao Chinese Academy of Medical Sciences & Peking Union Medical College Institute of Medicinal Plant Development Jingyuan Song ( [email protected] ) Research article Keywords: Corydalis saxicola, Corydalis tomentella, Papaveraceae, taxonomic study, cp genome Posted Date: April 17th, 2020 DOI: https://doi.org/10.21203/rs.3.rs-18411/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Version of Record: A version of this preprint was published at Ecology and Evolution on March 19th, 2021. See the published version at https://doi.org/10.1002/ece3.7312. Page 1/22 Abstract Backgroud: Corydalis DC., the largest genus of Papaveraceae, is recognized as one of the most taxonomically challenging plant taxa. However, no complete chloroplast (cp) genome for this genus has been reported to date. Results: We sequenced four complete cp genomes of two anities Corydalis saxicola and C. tomentellav of the genus Corydalis, compared these cp genomes with each other and others from Papaveraceae, and analyzed the phylogenetic relationships based on the sequences of common CDS. -

Bocconia and Macleaya Author(S): J
Bocconia and Macleaya Author(s): J. Hutchinson Source: Bulletin of Miscellaneous Information (Royal Botanic Gardens, Kew), Vol. 1920, No. 8 (1920), pp. 275-282 Published by: Springer on behalf of Royal Botanic Gardens, Kew Stable URL: http://www.jstor.org/stable/4120232 Accessed: 14-06-2016 14:47 UTC Your use of the JSTOR archive indicates your acceptance of the Terms & Conditions of Use, available at http://about.jstor.org/terms JSTOR is a not-for-profit service that helps scholars, researchers, and students discover, use, and build upon a wide range of content in a trusted digital archive. We use information technology and tools to increase productivity and facilitate new forms of scholarship. For more information about JSTOR, please contact [email protected]. Royal Botanic Gardens, Kew, Springer are collaborating with JSTOR to digitize, preserve and extend access to Bulletin of Miscellaneous Information (Royal Botanic Gardens, Kew) This content downloaded from 131.172.36.29 on Tue, 14 Jun 2016 14:47:17 UTC All use subject to http://about.jstor.org/terms 275 an old but no longer common species, 28 ft. high and 5 ft. in girth of trunk; and a fine healthy tree of the newer Japanese M. hypoleuca over 30 ft. high. There is a good collection of hardy bamboos on the lower, damper part of the garden, and it was interesting to see Phyllo- stachys aurea flowering, although not gratifying, since it portends the flowering and consequent death of the species throughout the country. XLIV.-BOCCONIA AND MACLEAYA. J. HUTCHINSON. The genus Bocconia (Papaveraceae) was founded by Linnaeus* in 1737, the type species being B. -

Influence of Different Elicitors on BIA Production in Macleaya Cordata
www.nature.com/scientificreports OPEN Infuence of diferent elicitors on BIA production in Macleaya cordata Peng Huang1,2,7, Liqiong Xia3,7, Li Zhou1,7, Wei Liu1,4, Peng Wang1, Zhixing Qing5* & Jianguo Zeng1,6* Sanguinarine (SAN) and chelerythrine (CHE) have been widely used as substitutes for antibiotics for decades. For a long time, SAN and CHE have been extracted from mainly Macleaya cordata, a plant species that is a traditional herb in China and belongs to the Papaveraceae family. However, with the sharp increase in demand for SAN and CHE, it is necessary to develop a new method to enhance the supply of raw materials. Here, we used methyl jasmonate (MJ), salicylic acid (SA) and wounding alone and in combination to stimulate aseptic seedlings of M. cordata at 0 h, 24 h, 72 h and 120 h and then compared the diferences in metabolic profles and gene expression. Ultimately, we found that the efect of using MJ alone was the best treatment, with the contents of SAN and CHE increasing by 10- and 14-fold, respectively. However, the increased SAN and CHE contents in response to combined wounding and MJ were less than those for induced by the treatment with MJ alone. Additionally, after MJ treatment, SAN and CHE biosynthetic pathway genes, such as those encoding the protopine 6-hydroxylase and dihydrobenzophenanthridine oxidase enzymes, were highly expressed, which is consistent with the accumulation of SAN and CHE. At the same time, we have also studied the changes in the content of synthetic intermediates of SAN and CHE after elicitor induction. -

J\V1ERICANI Iorticulturisf DECEMBER 1981
j\V1ERICANI IORTICULTURISf DECEMBER 1981 / lOmatoes in OCtober, November, and December? f course.. .and in January and February too with nutrients too. Your plants gradually absorb wnat they require Windowsill Gardens Nutriponics® Kits. If you simply and easily, eliminating the main causes of house O nave a sunny window you can grow tomatoes, plant failure; inadequate moisture and overwatering. peppers, geraniums, sunflowers, or whatever you wish in Nutriponics is a fully tested system. Over 65% of our our Gro-thru N Pots combined with our handsome planters customers reorder equipmemt and supplies, and kits are shown above. You can even grow your Nutriponic plants immediately available to get you started. Each includes our from seed. beautifully illustrated 50-page book on Nutriponics, to The lower container in this new system acts as a reservoir gether with Liqui-Soil, N Gro-thru Pots, and planting which enables the plant to take moisture as it needs it..and medium. Fill out the order coupon today. Windowsill Gardens, Grafton, N.H. 03240, Dept. AH D Senti Information Name, ___________~ =~_ D Send $4.95 kit Street'--______________ D Send $9.95 kit City State: _____Zip __ Include $2.00 for shipping WINDOWSILL GARDENS Grafton, New Hampshire 03240 #1ERICAN HORfICULTURIST DECEMBER 1981 FEATURES COLUMNS Camellias in Containers 16 Guest Editorial: Lessons We Can Learn Text and Photography by Anthony DeBlasi from Chelsea 2 A. St. Clair Wright Book Reviews 4 Gilbert S. Daniels Pronunciation Guide 8 Strange Relatives: The Cashew Family 9 Jane Steffey The Herb Garden: Thyme 13 Betty Ann Laws The Gotelli Dwarf Conifer Collection 20 Text by Steve Bender Photography by Barbara W. -

Downloaded from Protein Data Bank for Illustration in Ccp4mg
bioRxiv preprint doi: https://doi.org/10.1101/2020.10.12.333484; this version posted March 23, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Jurassic NLR: conserved and dynamic evolutionary features of the atypically ancient immune receptor ZAR1 Hiroaki Adachi1, Toshiyuki Sakai1, Jiorgos Kourelis1, Jose L. Gonzalez Hernandez2, Abbas Maqbool1 and Sophien Kamoun1* 1The Sainsbury Laboratory, University of East Anglia, Norwich Research Park, NR4 7UH, Norwich, UK 2Agronomy, Horticulture and Plant Sciences Department, South Dakota State University, Brookings, South Dakota, US *For correspondence: [email protected] ABSTRACT In plants, NLR immune receptors generally exhibit hallmarks of rapid evolution even at the intraspecific level. We reconstructed the evolutionary history of ZAR1, an atypically conserved NLR that traces its origin to early flowering plant lineages ~220 to 150 million years ago (Jurassic period). Ortholog sequence analyses revealed highly conserved features of ZAR1, including regions for pathogen effector recognition, intramolecular interactions and cell death activation. This uncovered a new conserved surface on the ZAR1 resistosome underside that is required for cell death induction. Throughout its evolution, ZAR1 acquired novel features, such as a C-terminal integration of a thioredoxin-like domain. ZAR1 duplicated into two paralog families, which underwent distinct evolutionary paths. We conclude that ZAR1 stands out among angiosperm NLRs for having experienced relatively limited gene duplication and expansion throughout its deep evolutionary history. Nonetheless, ZAR1 did also give rise to non-canonical NLR proteins with integrated domains and degenerated molecular features. -

The Botanic Garden
www.e-rara.ch The botanic garden Maund, Benjamin London, 1825-1836 ETH-Bibliothek Zürich Shelf Mark: Rar 1386 Persistent Link: http://dx.doi.org/10.3931/e-rara-16398 Chrysanthemum Sinense / Macleaya cordata / Leucocarpus alatus / Melittis melissophyllum. www.e-rara.ch Die Plattform e-rara.ch macht die in Schweizer Bibliotheken vorhandenen Drucke online verfügbar. Das Spektrum reicht von Büchern über Karten bis zu illustrierten Materialien – von den Anfängen des Buchdrucks bis ins 20. Jahrhundert. e-rara.ch provides online access to rare books available in Swiss libraries. The holdings extend from books and maps to illustrated material – from the beginnings of printing to the 20th century. e-rara.ch met en ligne des reproductions numériques d’imprimés conservés dans les bibliothèques de Suisse. L’éventail va des livres aux documents iconographiques en passant par les cartes – des débuts de l’imprimerie jusqu’au 20e siècle. e-rara.ch mette a disposizione in rete le edizioni antiche conservate nelle biblioteche svizzere. La collezione comprende libri, carte geografiche e materiale illustrato che risalgono agli inizi della tipografia fino ad arrivare al XX secolo. Nutzungsbedingungen Dieses Digitalisat kann kostenfrei heruntergeladen werden. Die Lizenzierungsart und die Nutzungsbedingungen sind individuell zu jedem Dokument in den Titelinformationen angegeben. Für weitere Informationen siehe auch [Link] Terms of Use This digital copy can be downloaded free of charge. The type of licensing and the terms of use are indicated in the title information for each document individually. For further information please refer to the terms of use on [Link] Conditions d'utilisation Ce document numérique peut être téléchargé gratuitement.