Federation of European Biological Systematic Societies Abstracts for Posters and Presentations
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Ecology of Soil Microarthropods in Gobi Gurvan Saykhan Mountains, Southern Mongolia Tsedev Bolortuya National University of Mongolia
University of Nebraska - Lincoln DigitalCommons@University of Nebraska - Lincoln Erforschung biologischer Ressourcen der Mongolei Institut für Biologie der Martin-Luther-Universität / Exploration into the Biological Resources of Halle-Wittenberg Mongolia, ISSN 0440-1298 2005 Ecology of Soil Microarthropods in Gobi Gurvan Saykhan Mountains, Southern Mongolia Tsedev Bolortuya National University of Mongolia Badamdorj Bayartogtokh National University of Mongolia, [email protected] Follow this and additional works at: http://digitalcommons.unl.edu/biolmongol Part of the Asian Studies Commons, Biodiversity Commons, Desert Ecology Commons, Environmental Sciences Commons, Nature and Society Relations Commons, Other Animal Sciences Commons, Terrestrial and Aquatic Ecology Commons, and the Zoology Commons Bolortuya, Tsedev and Bayartogtokh, Badamdorj, "Ecology of Soil Microarthropods in Gobi Gurvan Saykhan Mountains, Southern Mongolia" (2005). Erforschung biologischer Ressourcen der Mongolei / Exploration into the Biological Resources of Mongolia, ISSN 0440-1298. 120. http://digitalcommons.unl.edu/biolmongol/120 This Article is brought to you for free and open access by the Institut für Biologie der Martin-Luther-Universität Halle-Wittenberg at DigitalCommons@University of Nebraska - Lincoln. It has been accepted for inclusion in Erforschung biologischer Ressourcen der Mongolei / Exploration into the Biological Resources of Mongolia, ISSN 0440-1298 by an authorized administrator of DigitalCommons@University of Nebraska - Lincoln. In: Proceedings of the symposium ”Ecosystem Research in the Arid Environments of Central Asia: Results, Challenges, and Perspectives,” Ulaanbaatar, Mongolia, June 23-24, 2004. Erforschung biologischer Ressourcen der Mongolei (2005) 5. Copyright 2005, Martin-Luther-Universität. Used by permission. Erforsch. biol. Ress. Mongolei (Halle/Saale) 2005 (9): 53–58 Ecology of soil microarthropods in Gobi Gurvan Saykhan mountains, southern Mongolia Ts. -
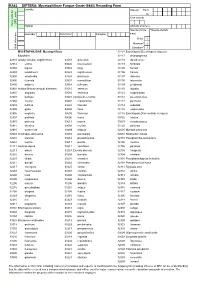
Ra82 Diptera
RA82 DIPTERA: Mycetophilinae Fungus Gnats (6480) Recording Form Locality Date(s) from: to: Vice county GPS users Grey cells for Habitat Altitude (metres) Source (circle *Source details Recorder Determiner Compiler one) Field 1 Museum* 2 Grid reference Literature* 3 MYCETOPHILIDAE: Mycetophilinae 33101 Exechiopsis (Exechiopsis) clypeata Exechiini 33117 dryaspagensis 32801 Allodia (Allodia) anglofennica 33519 griseolum33118 dumitrescae 32912 embla 33526 intermedium33119 fimbriata 32802 lugens 33522 kingi33120 furcata 32803 lundstroemi 33523 nigrofuscum33106 hammi 32804 ornaticollis 33524 proximum33107 indecisa 32806 truncata 33527 rosmellitum33108 intersecta 32805 zaitzevi 33514 ruficorne33109 jenkinsoni 32901 Allodia (Brachycampta) alternans 33515 serenum33110 ligulata 32910 angulata 33516 sericoma33121 magnicauda 32903 barbata 33601 Cordyla brevicornis33112 pseudindecisa 32904 czernyi 33602 crassicornis33113 pulchella 32909 foliifera 33603 fasciata33114 subulata 32905 grata 33604 fissa33115 unguiculata 32906 neglecta 33605 flaviceps33116 Exechiopsis (Xenexechia) crucigera 32907 pistillata 33606 fusca33002 leptura 32915 protenta 33613 insons33003 membranacea 32914 silvatica 33608 murina33122 pollicata 32916 westerholti 33609 nitidula32605 Myrosia maculosa 32602 Allodiopsis domestica 33610 parvipalpis32601 Notolopha cristata 32610 korolevi 33614 pseudomurina32701 Pseudexechia aurivernica 32607 rustica 33611 pusilla32706 monica 32212 Anatella alpina 33612 semiflava32705 parallela 32213 ankeli 33201 Exechia bicincta32703 trisignata 32216 bremia -

Major Clades of Agaricales: a Multilocus Phylogenetic Overview
Mycologia, 98(6), 2006, pp. 982–995. # 2006 by The Mycological Society of America, Lawrence, KS 66044-8897 Major clades of Agaricales: a multilocus phylogenetic overview P. Brandon Matheny1 Duur K. Aanen Judd M. Curtis Laboratory of Genetics, Arboretumlaan 4, 6703 BD, Biology Department, Clark University, 950 Main Street, Wageningen, The Netherlands Worcester, Massachusetts, 01610 Matthew DeNitis Vale´rie Hofstetter 127 Harrington Way, Worcester, Massachusetts 01604 Department of Biology, Box 90338, Duke University, Durham, North Carolina 27708 Graciela M. Daniele Instituto Multidisciplinario de Biologı´a Vegetal, M. Catherine Aime CONICET-Universidad Nacional de Co´rdoba, Casilla USDA-ARS, Systematic Botany and Mycology de Correo 495, 5000 Co´rdoba, Argentina Laboratory, Room 304, Building 011A, 10300 Baltimore Avenue, Beltsville, Maryland 20705-2350 Dennis E. Desjardin Department of Biology, San Francisco State University, Jean-Marc Moncalvo San Francisco, California 94132 Centre for Biodiversity and Conservation Biology, Royal Ontario Museum and Department of Botany, University Bradley R. Kropp of Toronto, Toronto, Ontario, M5S 2C6 Canada Department of Biology, Utah State University, Logan, Utah 84322 Zai-Wei Ge Zhu-Liang Yang Lorelei L. Norvell Kunming Institute of Botany, Chinese Academy of Pacific Northwest Mycology Service, 6720 NW Skyline Sciences, Kunming 650204, P.R. China Boulevard, Portland, Oregon 97229-1309 Jason C. Slot Andrew Parker Biology Department, Clark University, 950 Main Street, 127 Raven Way, Metaline Falls, Washington 99153- Worcester, Massachusetts, 01609 9720 Joseph F. Ammirati Else C. Vellinga University of Washington, Biology Department, Box Department of Plant and Microbial Biology, 111 355325, Seattle, Washington 98195 Koshland Hall, University of California, Berkeley, California 94720-3102 Timothy J. -

Ctenophore Relationships and Their Placement As the Sister Group to All Other Animals
ARTICLES DOI: 10.1038/s41559-017-0331-3 Ctenophore relationships and their placement as the sister group to all other animals Nathan V. Whelan 1,2*, Kevin M. Kocot3, Tatiana P. Moroz4, Krishanu Mukherjee4, Peter Williams4, Gustav Paulay5, Leonid L. Moroz 4,6* and Kenneth M. Halanych 1* Ctenophora, comprising approximately 200 described species, is an important lineage for understanding metazoan evolution and is of great ecological and economic importance. Ctenophore diversity includes species with unique colloblasts used for prey capture, smooth and striated muscles, benthic and pelagic lifestyles, and locomotion with ciliated paddles or muscular propul- sion. However, the ancestral states of traits are debated and relationships among many lineages are unresolved. Here, using 27 newly sequenced ctenophore transcriptomes, publicly available data and methods to control systematic error, we establish the placement of Ctenophora as the sister group to all other animals and refine the phylogenetic relationships within ctenophores. Molecular clock analyses suggest modern ctenophore diversity originated approximately 350 million years ago ± 88 million years, conflicting with previous hypotheses, which suggest it originated approximately 65 million years ago. We recover Euplokamis dunlapae—a species with striated muscles—as the sister lineage to other sampled ctenophores. Ancestral state reconstruction shows that the most recent common ancestor of extant ctenophores was pelagic, possessed tentacles, was bio- luminescent and did not have separate sexes. Our results imply at least two transitions from a pelagic to benthic lifestyle within Ctenophora, suggesting that such transitions were more common in animal diversification than previously thought. tenophores, or comb jellies, have successfully colonized from species across most of the known phylogenetic diversity of nearly every marine environment and can be key species in Ctenophora. -

Olympic Mushrooms 4/16/2021 Susan Mcdougall
Olympic Mushrooms 4/16/2021 Susan McDougall With links to species’ pages 206 species Family Scientific Name Common Name Agaricaceae Agaricus augustus Giant agaricus Agaricaceae Agaricus hondensis Felt-ringed Agaricus Agaricaceae Agaricus silvicola Forest Agaric Agaricaceae Chlorophyllum brunneum Shaggy Parasol Agaricaceae Chlorophyllum olivieri Olive Shaggy Parasol Agaricaceae Coprinus comatus Shaggy inkcap Agaricaceae Crucibulum laeve Common bird’s nest fungus Agaricaceae Cyathus striatus Fluted bird’s nest Agaricaceae Cystoderma amianthinum Pure Cystoderma Agaricaceae Cystoderma cf. gruberinum Agaricaceae Gymnopus acervatus Clustered Collybia Agaricaceae Gymnopus dryophilus Common Collybia Agaricaceae Gymnopus luxurians Agaricaceae Gymnopus peronatus Wood woolly-foot Agaricaceae Lepiota clypeolaria Shield dapperling Agaricaceae Lepiota magnispora Yellowfoot dapperling Agaricaceae Leucoagaricus leucothites White dapperling Agaricaceae Leucoagaricus rubrotinctus Red-eyed parasol Agaricaceae Morganella pyriformis Warted puffball Agaricaceae Nidula candida Jellied bird’s-nest fungus Agaricaceae Nidularia farcta Albatrellaceae Albatrellus avellaneus Amanitaceae Amanita augusta Yellow-veiled amanita Amanitaceae Amanita calyptroderma Ballen’s American Caesar Amanitaceae Amanita muscaria Fly agaric Amanitaceae Amanita pantheriana Panther cap Amanitaceae Amanita vaginata Grisette Auriscalpiaceae Lentinellus ursinus Bear lentinellus Bankeraceae Hydnellum aurantiacum Orange spine Bankeraceae Hydnellum complectipes Bankeraceae Hydnellum suaveolens -

CZECH MYCOLOGY Publication of the Czech Scientific Society for Mycology
CZECH MYCOLOGY Publication of the Czech Scientific Society for Mycology Volume 57 August 2005 Number 1-2 Central European genera of the Boletaceae and Suillaceae, with notes on their anatomical characters Jo s e f Š u t a r a Prosetická 239, 415 01 Tbplice, Czech Republic Šutara J. (2005): Central European genera of the Boletaceae and Suillaceae, with notes on their anatomical characters. - Czech Mycol. 57: 1-50. A taxonomic survey of Central European genera of the families Boletaceae and Suillaceae with tubular hymenophores, including the lamellate Phylloporus, is presented. Questions concerning the delimitation of the bolete genera are discussed. Descriptions and keys to the families and genera are based predominantly on anatomical characters of the carpophores. Attention is also paid to peripheral layers of stipe tissue, whose anatomical structure has not been sufficiently studied. The study of these layers, above all of the caulohymenium and the lateral stipe stratum, can provide information important for a better understanding of relationships between taxonomic groups in these families. The presence (or absence) of the caulohymenium with spore-bearing caulobasidia on the stipe surface is here considered as a significant ge neric character of boletes. A new combination, Pseudoboletus astraeicola (Imazeki) Šutara, is proposed. Key words: Boletaceae, Suillaceae, generic taxonomy, anatomical characters. Šutara J. (2005): Středoevropské rody čeledí Boletaceae a Suillaceae, s poznámka mi k jejich anatomickým znakům. - Czech Mycol. 57: 1-50. Je předložen taxonomický přehled středoevropských rodů čeledí Boletaceae a. SuiUaceae s rourko- vitým hymenoforem, včetně rodu Phylloporus s lupeny. Jsou diskutovány otázky týkající se vymezení hřibovitých rodů. Popisy a klíče k čeledím a rodům jsou založeny převážně na anatomických znacích plodnic. -

Identity of Mertensia Oblongifolia (Nutt.) G. Don (Boraginaceae) and Its Allies in Western North America
Great Basin Naturalist Volume 58 Number 1 Article 4 1-30-1998 Identity of Mertensia oblongifolia (Nutt.) G. Don (Boraginaceae) and its allies in western North America Ahmed M. Warfa Brigham Young University Follow this and additional works at: https://scholarsarchive.byu.edu/gbn Recommended Citation Warfa, Ahmed M. (1998) "Identity of Mertensia oblongifolia (Nutt.) G. Don (Boraginaceae) and its allies in western North America," Great Basin Naturalist: Vol. 58 : No. 1 , Article 4. Available at: https://scholarsarchive.byu.edu/gbn/vol58/iss1/4 This Article is brought to you for free and open access by the Western North American Naturalist Publications at BYU ScholarsArchive. It has been accepted for inclusion in Great Basin Naturalist by an authorized editor of BYU ScholarsArchive. For more information, please contact [email protected], [email protected]. Great Basin Naturalist 58(1), © 1998, pp. 38-44 IDENTITY OF MERTENSIA OBLONGIFOLIA (NUTT) G. DON (BORAGINACEAE) AND ITS ALLIES IN WESTERN NORTH AMERICA Ahmed M. Warfal ABSTRACT.-The current status of Mertensia oblongifoUa (Nutt.) G. Don and its allied taxa is surveyed. On the bases of continuously coherent morphological characters and/or regionally correlated variations, more than 30 taxa, including species, subspecies, varieties, and 1 forma, previously considered different from M. oblongifolia, are now placed under synonymy of this species. Those taxa currently known as M. fusiformis Greene, M. bakeri Greene, and M. bakeri var. osterhoutii Williams are among the new synonyms. Typification, taxonomy, and morphological problems ofM. oblongifo lia are discussed. Key words: Mertensia oblongifolia, typification, Wxorwmy, morphology, allied taxa. Nuttall (1834) described and depicted Pul represents an important additional morpholog monaria oblongifolia from a collection ofplants ical feature in the taxon. -

Prickly News South Coast Cactus & Succulent Society Newsletter | Feb 2021
PRICKLY NEWS SOUTH COAST CACTUS & SUCCULENT SOCIETY NEWSLETTER | FEB 2021 Guillermo ZOOM PRESENTATION SHARE YOUR GARDEN OR YOUR FAVORITE PLANT Rivera Sunday, February 14 @ 1:30 pm Cactus diversity in northwestern Argentina: a habitat approach I enjoyed Brian Kemble’s presentation on the Ruth Bancroft Garden in Walnut Creek. For those of you who missed the presentation, check out the website at https://www. ruthbancroftgarden.org for hints on growing, lectures and access to webinars that are available. Email me with photos of your garden and/or plants Brian graciously offered to answer any questions that we can publish as a way of staying connected. or inquiries on the garden by contacting him at [email protected] [email protected]. CALL FOR PHOTOS: The Mini Show genera for February are Cactus: Eriosyce (includes Neoporteria, Islaya and Neochilenia) and Succulent: Crassula. Photos will be published and you will be given To learn more visit southcoastcss.org one Mini-show point each for a submitted photo of your cactus, succulent or garden (up to 2 points). Please include your plant’s full name if you know it (and if you don’t, I will seek advice for you). Like us on our facebook page Let me know if you would prefer not to have your name published with the photos. The photos should be as high resolution as possible so they will publish well and should show off the plant as you would Follow us on Instagram, _sccss_ in a Mini Show. This will provide all of us with an opportunity to learn from one another and share plants and gardens. -

Hábitats De Turbera En La Red Natura 2000 Diagnosis Y Criterios Para Su Conservación Y Gestión En La Región Biogeográfica Atlántica
Hábitats de turbera en la Red Natura 2000 Diagnosis y criterios para su conservación y gestión en la Región Biogeográfica Atlántica Pablo Ramil-Rego Manuel A. Rodríguez Guitián (Editores) Hábitats de turbera en la Red Natura 2000 Diagnosis y criterios para su conservación y gestión en la Región Biogeográfica Atlántica Pablo Ramil-Rego - Manuel A. Rodríguez Guitián (Eds.) Lugo 2017 Título: Hábitats de turbera en la Red Natura 2000. Diagnosis y criterios para su conservación y gestión en la Región Biogeográfica Atlántica Editores: Pablo Ramil-Rego, Manuel A. Rodríguez Guitián A efectos bibliográficos a obra debe citarse: Obra Completa: Ramil-Rego, P, Rodríguez Guitián M.A. (Eds.) (2017). Hábitats de turbera en la Red Natura 2000. Diag- nosis y criterios para su conservación y gestión en la Región Biogeográfica Atlántica. Horreum-Ibader, Lugo. 427p. Capítulo concreto: Ramil-Rego, P., López Castro, H., Muñoz Sobrino. C., Rodríguez Guitián, M.A., Gómez Orellana, L., Ferreiro Da Costa, J. (2017). Información Territorial: Unión Europea. En: Ramil-Rego, P, Rodríguez Guitián M.A. (Eds.), Hábitats de turbera en la Red Natura 2000. Diagnosis y criterios para su conservación y gestión en la Región Biogeográfica Atlántica: 149-190. Horreum-Ibader, Lugo. Esta publicación foi sometida a un proceso de revisión por pares. Edita: Horreum - IBADER Copyright: IBADER - Horreum A totalidade dos textos, gráficos e imaxes publicadas nesta obra están protexidos por copyright. Queda prohibida a repro- dución total ou parcial por calquera medio gráfico ou electrónico -

Chemical Elements in Ascomycetes and Basidiomycetes
Chemical elements in Ascomycetes and Basidiomycetes The reference mushrooms as instruments for investigating bioindication and biodiversity Roberto Cenci, Luigi Cocchi, Orlando Petrini, Fabrizio Sena, Carmine Siniscalco, Luciano Vescovi Editors: R. M. Cenci and F. Sena EUR 24415 EN 2011 1 The mission of the JRC-IES is to provide scientific-technical support to the European Union’s policies for the protection and sustainable development of the European and global environment. European Commission Joint Research Centre Institute for Environment and Sustainability Via E.Fermi, 2749 I-21027 Ispra (VA) Italy Legal Notice Neither the European Commission nor any person acting on behalf of the Commission is responsible for the use which might be made of this publication. Europe Direct is a service to help you find answers to your questions about the European Union Freephone number (*): 00 800 6 7 8 9 10 11 (*) Certain mobile telephone operators do not allow access to 00 800 numbers or these calls may be billed. A great deal of additional information on the European Union is available on the Internet. It can be accessed through the Europa server http://europa.eu/ JRC Catalogue number: LB-NA-24415-EN-C Editors: R. M. Cenci and F. Sena JRC65050 EUR 24415 EN ISBN 978-92-79-20395-4 ISSN 1018-5593 doi:10.2788/22228 Luxembourg: Publications Office of the European Union Translation: Dr. Luca Umidi © European Union, 2011 Reproduction is authorised provided the source is acknowledged Printed in Italy 2 Attached to this document is a CD containing: • A PDF copy of this document • Information regarding the soil and mushroom sampling site locations • Analytical data (ca, 300,000) on total samples of soils and mushrooms analysed (ca, 10,000) • The descriptive statistics for all genera and species analysed • Maps showing the distribution of concentrations of inorganic elements in mushrooms • Maps showing the distribution of concentrations of inorganic elements in soils 3 Contact information: Address: Roberto M. -

Albuca Spiralis
Flowering Plants of Africa A magazine containing colour plates with descriptions of flowering plants of Africa and neighbouring islands Edited by G. Germishuizen with assistance of E. du Plessis and G.S. Condy Volume 62 Pretoria 2011 Editorial Board A. Nicholas University of KwaZulu-Natal, Durban, RSA D.A. Snijman South African National Biodiversity Institute, Cape Town, RSA Referees and other co-workers on this volume H.J. Beentje, Royal Botanic Gardens, Kew, UK D. Bridson, Royal Botanic Gardens, Kew, UK P. Burgoyne, South African National Biodiversity Institute, Pretoria, RSA J.E. Burrows, Buffelskloof Nature Reserve & Herbarium, Lydenburg, RSA C.L. Craib, Bryanston, RSA G.D. Duncan, South African National Biodiversity Institute, Cape Town, RSA E. Figueiredo, Department of Plant Science, University of Pretoria, Pretoria, RSA H.F. Glen, South African National Biodiversity Institute, Durban, RSA P. Goldblatt, Missouri Botanical Garden, St Louis, Missouri, USA G. Goodman-Cron, School of Animal, Plant and Environmental Sciences, University of the Witwatersrand, Johannesburg, RSA D.J. Goyder, Royal Botanic Gardens, Kew, UK A. Grobler, South African National Biodiversity Institute, Pretoria, RSA R.R. Klopper, South African National Biodiversity Institute, Pretoria, RSA J. Lavranos, Loulé, Portugal S. Liede-Schumann, Department of Plant Systematics, University of Bayreuth, Bayreuth, Germany J.C. Manning, South African National Biodiversity Institute, Cape Town, RSA A. Nicholas, University of KwaZulu-Natal, Durban, RSA R.B. Nordenstam, Swedish Museum of Natural History, Stockholm, Sweden B.D. Schrire, Royal Botanic Gardens, Kew, UK P. Silveira, University of Aveiro, Aveiro, Portugal H. Steyn, South African National Biodiversity Institute, Pretoria, RSA P. Tilney, University of Johannesburg, Johannesburg, RSA E.J. -

Zootaxa, Diptera, Mycetophilidae
Zootaxa 856: 1–35 (2005) ISSN 1175-5326 (print edition) www.mapress.com/zootaxa/ ZOOTAXA 856 Copyright © 2005 Magnolia Press ISSN 1175-5334 (online edition) A review of fungus gnats in the tribe Exechiini (Diptera, Myceto- philidae) from the J. W. Zetterstedt collection at the Museum of Zoology in Lund, Sweden JOSTEIN KJÆRANDSEN Museum of Zoology, Lund University, Helgonavägen 3, S-223 62 Lund, Sweden. ([email protected]) Table of contents Abstract . 1 Introduction . 2 Material and methods . 4 Result . 6 List of species . 12 Acknowledgements . 33 References . 33 Abstract The collections of fungus gnats by Johan Wilhelm Zetterstedt (1785–1874), lodged in the Museum of Zoology in Lund, Sweden, are examined for all species belonging in the tribe Exechiini Edwards. The majority of the material was collected in Fennoscandia, mainly in Sweden, in the first half of the 19th century. Altogether 37 species of the tribe Exechiini could be safely identified. Three additional species are strongly indicated to be present in the collections, but could not be identified with certainty, viz. Allodia (Brachycampta) alternans (Zetterstedt, 1838), Cordyla murina Winnertz, 1863 and Stigmatomeria crassicornis (Stannius, 1831). Some of Zetterstedt's types have been erroneously synonymized and misinterpreted in modern literature. Hence, illustra- tions of terminalia are presented for all recognizable Exechiini types described by Zetterstedt. In order to preserve nomenclatural stability a lectotype is selected for Brevicornu griseolum (Zetter- stedt, 1852) sensu auctore nec Edwards, and a neotype is selected for Allodia (Brachycampta) alternans (Zetterstedt, 1838). Two species names are reinstated, viz. Brevicornu canescens (Zetter- stedt, 1852) sp.