Distillation 65 Chem 355 Jasperse DISTILLATION
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Extraction of Wax from Sorghum Bran •
i EXTRACTION OF WAX FROM SORGHUM BRAN • 1 HSIEN-WEN HSU <& . ***£/ B. S., National Taiwan University, China, 1951 A THESIS » submitted in partial fulfillment of the requirements for the degree MASTER OF SCIENCE - Department of Chemical Engineering KANSAS STATE COLLEGE OF AGRICULTURE AND APPLIED SCIENCE - 1955 » k Lo i~i ii Q^cu^v^Js TABLE OF CONTENTS INTRODUCTION 1 PREVIOUS WORK 3 MATERIALS 6 GLASS SOXHLET EXTRACTION AND ANALYSIS OF WAX AND OIL IN MISCELLA 7 Procedure of Using Soxhlet Extractor 7 Separation of Wax from Oil by the Acetone Method.. 8 Summary of Results 9 PILOT PLANT EXTRACTION 15 SEPARATION OF WAX AND OIL 16 Effect of Temperature in the Acetone Method 16 Urea Complex Method of Separation. 17 Theory 17 Experimental Procedure 22 Summary of Results 26 COST ESTIMATION OF A WAX RECOVERY PLANT 29 DISCUSSION 30 CONCLUSIONS 35 ACKNOWLEDGMENTS 36 BIBLIOGRAPHY 37 APPENDIX 39 INTRODUCTION i One of the functions of the Agricultural Experiment Station is the never ending search for means of better and fuller utilization of the agri- cultural products that are of particular interest in the state of Kansas. In line with this broad objective, the extraction of economically valuable lipid materials from the sorghum bran has been studied in several theses in the Departments of Chemical Engineering and Chemistry in recent years. Generally the emphasis has b. en placed upon either the chemical and physical properties of these lipides or the equipment design and performance for the solvent ex- traction operation. A brief review of the previous work is presented in the ensuing section. -

The Separation of Three Azeotropes by Extractive Distillation by An-I Yeh A
The separation of three azeotropes by extractive distillation by An-I Yeh A thesis submitted in partial fulfillment of the requirement for the degree of Master of Science in Chemical Engineering Montana State University © Copyright by An-I Yeh (1983) Abstract: Several different kinds of extractive distillation agents were investigated to affect the separation of three binary liquid mixtures, isopropyl ether - acetone, methyl acetate - methanol, and isopropyl ether - methyl ethyl ketone. Because of the small size of the extractive distillation column, relative volatilities were assumed constant and the Fenske equation was used to calculate the relative volatilities and the number of minimum theoretical plates. Dimethyl sulfoxide was found to be a good extractive distillation agent. Extractive distillation when employing a proper agent not only negated the azeotropes of the above mixtures, but also improved the efficiency of separation. This process could reverse the relative volatility of isopropyl ether and acetone. This reversion was also found in the system of methyl acetate and methanol when nitrobenzene was the agent. However, normal distillation curves were obtained for the system of isopropyl ether and methyl ethyl ketone undergoing extractive distillation. In the system of methyl acetate and methanol, the relative volatility decreased as the agents' carbon number increased when glycols were used as the agents. In addition, the oxygen number and the locations of hydroxyl groups in the glycols used were believed to affect the values of relative volatility. An appreciable amount of agent must be maintained in the column to affect separation. When dimethyl sulfoxide was an agent for the three systems studied, the relative volatility increased as the addition rate increased. -
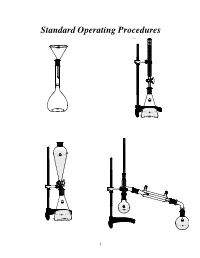
Standard Operating Procedures
Standard Operating Procedures 1 Standard Operating Procedures OVERVIEW In the following laboratory exercises you will be introduced to some of the glassware and tech- niques used by chemists to isolate components from natural or synthetic mixtures and to purify the individual compounds and characterize them by determining some of their physical proper- ties. While working collaboratively with your group members you will become acquainted with: a) Volumetric glassware b) Liquid-liquid extraction apparatus c) Distillation apparatus OBJECTIVES After finishing these sessions and reporting your results to your mentor, you should be able to: • Prepare solutions of exact concentrations • Separate liquid-liquid mixtures • Purify compounds by recrystallization • Separate mixtures by simple and fractional distillation 2 EXPERIMENT 1 Glassware Calibration, Primary and Secondary Standards, and Manual Titrations PART 1. Volumetric Glassware Calibration Volumetric glassware is used to either contain or deliver liquids at a specified temperature. Glassware manufacturers indicate this by inscribing on the volumetric ware the initials TC (to contain) or TD (to deliver) along with the calibration temperature, which is usually 20°C1. Volumetric glassware must be scrupulously clean before use. The presence of streaks or droplets is an indication of the presence of a grease film. To eliminate grease from glassware, scrub with detergent solution, rinse with tap water, and finally rinse with a small portion of distilled water. Volumetric flasks (TC) A volumetric flask has a large round bottom with only one graduation mark positioned on the long narrow neck. Graduation Mark Stopper The position of the mark facilitates the accurate and precise reading of the meniscus. If the flask is used to prepare a solution starting with a solid compound, add small amounts of sol- vent until the entire solid dissolves. -

Experiment 2 — Distillation and Gas Chromatography
Chem 21 Fall 2009 Experiment 2 — Distillation and Gas Chromatography _____________________________________________________________________________ Pre-lab preparation (1) Read the supplemental material on distillation theory and techniques from Zubrick, The Organic Chem Lab Survival Manual, and the section on Gas Chromatography from Fessenden, Fessenden, and Feist, Organic Laboratory Techniques, then read this handout carefully. (2) In your notebook, write a short paragraph summarizing what you will be doing in this experiment and what you hope to learn about the efficiencies of the distillation techniques. (3) Sketch the apparatus for the simple and fractional distillations. Your set-up will look much like that shown on p 198 of Zubrick, except that yours will have a simple drip tip in place of the more standard vacuum adaptor. (4) Look up the structures and relevant physical data for the two compounds you will be using. What data are relevant? Read the procedure, think about the data analysis, and decide what you need. (5) Since you have the necessary data, calculate the log of the volatility factor (log α) that you will need for the theoretical plate calculation. Distillation has been used since antiquity to separate the components of mixtures. In one form or another, distillation is used in the manufacture of perfumes, flavorings, liquors, and a variety of other organic chemicals. One of its most important modern applications is in refining crude oil to make fuels, lubricants, and other petrochemicals. The first step in the refining process is separation of crude petroleum into various hydrocarbon fractions by distillation through huge fractionating columns, called distillation towers, that are hundreds of feet high. -

Laboratory Rectifying Stills of Glass 2
» LABORATORY RECTIFYING STILLS OF GLASS 2 By Johannes H. Bruun 3 and Sylvester T. Schicktanz 3 ABSTRACT A complete description is given of a set of all-glass rectifying stills, suitable for distillation at pressures ranging from atmospheric down to about 50 mm. The stills are provided with efficient bubbling-cap columns containing 30 to 60 plates. Adiabatic conditions around the column are maintained by surrounding it with a jacket provided with a series of independent electrical heating units. Suitable means are provided for adjusting or maintaining the reflux ratio at the top of the column. For the purpose of conveniently obtaining an accurate value of the true boiling points of the distillates a continuous boiling-point apparatus is incor- porated in the receiving system. An efficient still of the packed-column type for distillations under pressures less than 50 mm is also described. Methods of operation and efficiency tests are given for the stills. CONTENTS Page I. Introduction 852 II. General features of the still assembly 852 III. Still pot 853 IV. Filling tube and heater for the still pot 853 V. Rectifying column 856 1. General features 856 2. Large-size column 856 3. Medium-size column 858 4. Small-size column 858 VI. Column jacket 861 VII. Reflux regulator 861 1. With variable reflux ratio 861 2. With constant reflux ratio 863 VIII. The continuous boiling-point apparatus 863 IX. Condensers and receiver 867 X. The mounting of the still 867 XI. Manufacture and transportation of the bubbling-cap still 870 XII. Operation of the bubbling-cap still 870 XIII. -

Fractional Distillation (Ideal) 1-Propanol/2-Propanol at 760 Torr
Statistical Molecular Thermodynamics Christopher J. Cramer Video 10.7 Azeotropes and Immiscible Phases Fractional Distillation (Ideal) 1-propanol/2-propanol at 760 torr (from Video 10.5) bp as a function of composition x1−propanol(Solution) y1−propanol (Vapor) At 90 °C (between the two pure component boiling points) € the composition of the liquid is 59 mol% 1-propanol (point a) and the vapor is 45 mol% 1-propanol (point b) Fractional distillation: vapor is condensed and revaporized many times a to b to c to d to ... and finally you get to the pure lower boiling component at the top (the coolest point) of a distillation head! Fractional Distillation (Non-Ideal) benzene/ethanol at 760 torr Using Dalton’s/Raoult’s laws: * P2 x2P2 b b y = = a a 2 760 torr lim x2→1 760 torr d c d c and Dalton’s/Henry’s laws: f e e P2 x2kH,2 y2 = = 760 torr lim x2→0 760 torr bp as a function of composition Deviation from ideality permits the vapor phase to be enriched in either component depending on liquid composition. Distillation of benzene-rich mixture Distillation of ethanol-rich mixture Fractional distillation: vapor is condensed and revaporized many times a to b to c to d to e to f, but, irrespective of initial composition, ultimately an azeotropic composition is reached (about 55:45 above) that cannot be further separated by distillation! Self-assessment What purification can be accomplished for a liquid mixture that is not already at the azeotropic composition? Self-assessment Explained If not already at the azeotropic composition, the azeotrope at the top of b b a a the distillation head will be carrying away a greater fraction of one component than d is present in the liquid. -

Crude Oil Distillation
fact sheet Crude oil distillation Crude oil is a complex mixture of hydrocarbon molecules of all different shapes and lengths. This rich soup is where we get petrol for our cars, bitumen for our roads, and oils for our engines, amongst many other things. To create any of the thousands of useful things made from oil different hydrocarbons have to be separated. methane This is done by separating or refining using a distillation process. Hydrocarbons Hydrocarbons are molecules made from carbon and hydrogen atoms. With a chain of carbon as their ‘backbone’, hydrocarbons differ in their length and can form chains, branches or even rings. This backbone can range from just one carbon atom in length to more than 60 atoms. cyclohexane Methane has a single carbon atom; cyclohexane has six carbon atoms in a ring; and octane has eight carbon atoms in a chain. Even slight differences in length and structure of hydrocarbon molecules can give different chemical and physical properties. The refining process separates molecules according to their different boiling points. Hydrocarbons are jumbled together as thick black crude oil. The most efficient way to separate them into smaller fractions is by a process called fractional distillation. octane Hydrocarbons are found as solids, liquids and gases based on their length Short hydrocarbons (one to A backbone of five or more carbons Once a hydrocarbon gets so four carbons) are gases at room will give you a liquid hydrocarbon. large and cumbersome that it temperatures because they have Shorter liquid hydrocarbons will boil has a backbone of more than 20 such low boiling points. -

Distillation1
Distillation1 Distillation is a commonly used method for purifying liquids and separating mixtures of liquids into their individual components. Familiar examples include the distillation of crude fermentation broths into alcoholic spirits such as gin and vodka, and the fractionation of crude oil into useful products such as gasoline and heating oil. In the organic lab, distillation is used for purifying solvents and liquid reaction products. In analyzing a distillation, how do we know the real composition of each collected component? In this lab, we will introduce gas chromatography (GC), which will tell us how pure each fraction we collected is. After the distillation of your unknown is complete, you will analyze both components via GC. See page 11 and 12 for a light discussion on GC. To understand distillation, first consider what happens upon heating a liquid. At any temperature, some molecules of a liquid possess enough kinetic energy to escape into the vapor phase (evaporation) and some of the molecules in the vapor phase return to the liquid (condensation). An equilibrium is set up, with molecules going back and forth between liquid and vapor. At higher temperatures, more molecules possess enough kinetic energy to escape, which results in a greater number of molecules being present in the vapor phase. If the liquid is placed into a closed container with a pressure gauge attached, one can obtain a quantitative measure of the degree of vaporization. This pressure is defined as the vapor pressure of the compound, which can be measured at different temperatures. Consider heating cyclohexane, a liquid hydrocarbon, and measuring its vapor pressure at different temperatures. -

CHEM 344 Distillation of Liquid Mixtures
CHEM 344 Distillation of liquid mixtures 1. Distillation basics The vaporization of a liquid and condensation of the resulting vapor is the basis of distillation. Organic liquids containing small amounts (<15%) of impurities or non-volatile substances are easily purified by simple distillation, as are liquid mixtures where the difference in boiling point of the components is >70 oC. Fractional distillation is more useful for separating mixtures of liquids where the boiling points of the components differ by <70 oC (see later). A typical simple distillation setup is shown in Figure 1. It consists of a flask containing the liquid to be distilled, an adapter to hold a thermometer and to connect the flask to a water-cooled condenser, and a flask to hold the condensed liquid (the distillate). Figure 1: Apparatus for a simple distillation. 1.1 The distillation flask The distillation flask is a round-bottom flask. The liquid to be distilled should fill the distillation flask to ~50-60% of its capacity. To promote even heating of the liquid, a boiling chip or a magnetic stir bar is added before heat is applied to the distillation flask. The irregular chips provide sites for bubbles of vapor to form, or alternatively the liquid is agitated with the magnetic stirrer as it is being heated. Never add a boiling chip or a stir bar to a hot liquid! Doing so can cause a seemingly calm liquid to boil suddenly and violently. 1 1.2 The distilling adapter The adapter connects the distillation flask, the condenser, and the thermometer. This type of adapter is often referred to as a distillation head. -

Fractional Distillation for the Laboratory and Industry
ll distillations attempt to separate a lower boiling material (A) from a higher boiling material (B). Packed columns are “packed” A with a material that creates a large surface area within the distillation column. Vapors rise through the column, are condensed by the condenser and fall back down the column. This material is called the condensate. The descending condensate “wets” the packing. Ascending vapors rise through the wet packing and are forced into intimate contact with the condensate. This intimate contact causes the vapor to become enriched in the lower boiling material. This process is often called rectification. A A A AA AA A AA A A AA A A A AA A AA A B/R Instrument Corporation B/R Instrument was founded in 1966 by Roger Roark, Sr. and B A A A A A A B B Harry Brown, Jr. Originally a manufacturer of general glass- A A A AA A A B A A AA AA ware, the company began to specialize in the manufacture B B A A A A A AA AA A A A of spinning band distillation systems in 1968 under a DuPont B A B B B B patent. Since then the company has grown steadily in the A A A A distillation and laboratory equipment markets and now A B A B A B A A A AA distributes products to customers throughout the world. A B A A AA A A A B B A AA A Located in Easton, Maryland on the beautiful Eastern B A A A A A A A B A AA B A Shore of the Chesapeake Bay, the B/R facility encompasses A AA © 2004, B/R Instrument Corporation B A B AA A 14,700 square feet (1,370 square meters) and houses manu- B B A B B A B B A A B A A B A A B A A facturing, sales, engineering, service and administrative A A A A A B B A divisions. -

A Large Spinning-Band Fractionating Column for Use with Small Quantities of Liquids
468 INDUSTRIAL AND ENGINEERING CHEMISTRY VOL. 12, h-0.8 Operation of Viscometer After a viscosity measurement has been made, the liquid The filling operation is effected by means of the filling tube may be removed readily by applying a vacuum to the leveling illustrated in Figure 3. The asphaltic liquid is warmed to a tube. Benzene or carbon tetrachloride may be introduced temperature (60' to 82.22' C., 140" to 180OF.) sufficient to into the left arm, and as it is sucked through the instrument it ymit its being poured into the 100-mesh copper funnel sieve. he liquid passes through the sieve into the receiver arm of the sweeps the viscometer clean of oil. As the vacuum is con- viscometer and catches at point P where the narrow connecting tinued, the cleaning solvent is soon evaporated. The cleaning tube joins the receiver tube reservoir. The flow down through operation is thus effected simply without removal of the the connecting tubing is controlled by means of pressure changes viscometer from the thermostat bath. caused by a roller device (devised by V. Lantz, of these labora- tories, and shown in Figure 2) which squeezes a rubber tubing The viscometer is readily calibrated by the use of oils of connected to the left arm of the viscometer. To control the rate known viscosity (such as the alpha and beta oils of the Ameri- of flow from the filling tube into the viscometer and to adjust can Petroleum Institute) at low enough temperatures to get the flow so that the upper surface of the liquid approaches mark good flow times. -

Distillation Strategies: a Key Factor to Obtain Spirits with Specific Organoleptic Characteristics
DISTILLATION STRATEGIES: A KEY FACTOR TO OBTAIN SPIRITS WITH SPECIFIC ORGANOLEPTIC CHARACTERISTICS Pau Matias-Guiu Martí ADVERTIMENT. L'accés als continguts d'aquesta tesi doctoral i la seva utilització ha de respectar els drets de la persona autora. Pot ser utilitzada per a consulta o estudi personal, així com en activitats o materials d'investigació i docència en els termes establerts a l'art. 32 del Text Refós de la Llei de Propietat Intel·lectual (RDL 1/1996). Per altres utilitzacions es requereix l'autorització prèvia i expressa de la persona autora. En qualsevol cas, en la utilització dels seus continguts caldrà indicar de forma clara el nom i cognoms de la persona autora i el títol de la tesi doctoral. No s'autoritza la seva reproducció o altres formes d'explotació efectuades amb finalitats de lucre ni la seva comunicació pública des d'un lloc aliè al servei TDX. Tampoc s'autoritza la presentació del seu contingut en una finestra o marc aliè a TDX (framing). Aquesta reserva de drets afecta tant als continguts de la tesi com als seus resums i índexs. ADVERTENCIA. El acceso a los contenidos de esta tesis doctoral y su utilización debe respetar los derechos de la persona autora. Puede ser utilizada para consulta o estudio personal, así como en actividades o materiales de investigación y docencia en los términos establecidos en el art. 32 del Texto Refundido de la Ley de Propiedad Intelectual (RDL 1/1996). Para otros usos se requiere la autorización previa y expresa de la persona autora. En cualquier caso, en la utilización de sus contenidos se deberá indicar de forma clara el nombre y apellidos de la persona autora y el título de la tesis doctoral.