Could the Xenon Dioxide Molecule Exist?† Pekka Pyykk, and Toomas Tamm J
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Potential Ivvs.Gelbasedre
US010644304B2 ( 12 ) United States Patent ( 10 ) Patent No.: US 10,644,304 B2 Ein - Eli et al. (45 ) Date of Patent : May 5 , 2020 (54 ) METHOD FOR PASSIVE METAL (58 ) Field of Classification Search ACTIVATION AND USES THEREOF ??? C25D 5/54 ; C25D 3/665 ; C25D 5/34 ; ( 71) Applicant: Technion Research & Development HO1M 4/134 ; HOTM 4/366 ; HO1M 4/628 ; Foundation Limited , Haifa ( IL ) (Continued ) ( 72 ) Inventors : Yair Ein - Eli , Haifa ( IL ) ; Danny (56 ) References Cited Gelman , Haifa ( IL ) ; Boris Shvartsev , Haifa ( IL ) ; Alexander Kraytsberg , U.S. PATENT DOCUMENTS Yokneam ( IL ) 3,635,765 A 1/1972 Greenberg 3,650,834 A 3/1972 Buzzelli ( 73 ) Assignee : Technion Research & Development Foundation Limited , Haifa ( IL ) (Continued ) ( * ) Notice : Subject to any disclaimer , the term of this FOREIGN PATENT DOCUMENTS patent is extended or adjusted under 35 CN 1408031 4/2003 U.S.C. 154 ( b ) by 56 days . EP 1983078 10/2008 (21 ) Appl. No .: 15 /300,359 ( Continued ) ( 22 ) PCT Filed : Mar. 31 , 2015 OTHER PUBLICATIONS (86 ) PCT No .: PCT/ IL2015 /050350 Hagiwara et al. in ( Acidic 1 - ethyl - 3 -methylimidazoliuum fluoride: a new room temperature ionic liquid in Journal of Fluorine Chem $ 371 (c ) ( 1 ), istry vol . 99 ( 1999 ) p . 1-3 ; ( Year: 1999 ). * (2 ) Date : Sep. 29 , 2016 (Continued ) (87 ) PCT Pub . No .: WO2015 / 151099 Primary Examiner — Jonathan G Jelsma PCT Pub . Date : Oct. 8 , 2015 Assistant Examiner Omar M Kekia (65 ) Prior Publication Data (57 ) ABSTRACT US 2017/0179464 A1 Jun . 22 , 2017 Disclosed is a method for activating a surface of metals , Related U.S. Application Data such as self- passivated metals , and of metal -oxide dissolu tion , effected using a fluoroanion -containing composition . -

Chemistry of the Noble Gases*
CHEMISTRY OF THE NOBLE GASES* By Professor K. K. GREE~woon , :.\I.Sc., sc.D .. r".lU.C. University of N ewca.stle 1tpon Tyne The inert gases, or noble gases as they are elements were unsuccessful, and for over now more appropriately called, are a remark 60 years they epitomized chemical inertness. able group of elements. The lightest, helium, Indeed, their electron configuration, s2p6, was recognized in the gases of the sun before became known as 'the stable octet,' and this it was isolated on ea.rth as its name (i]A.tos) fotmed the basis of the fit·st electronic theory implies. The first inert gas was isolated in of valency in 1916. Despite this, many 1895 by Ramsay and Rayleigh; it was named people felt that it should be possible to induce argon (apy6s, inert) and occurs to the extent the inert gases to form compounds, and many of 0·93% in the earth's atmosphere. The of the early experiments directed to this end other gases were all isolated before the turn have recently been reviewed.l of the century and were named neon (v€ov, There were several reasons why chemists new), krypton (KpVn'TOV, hidden), xenon believed that the inert gases might form ~€vov, stmnger) and radon (radioactive chemical compounds under the correct con emanation). Though they occur much less ditions. For example, the ionization poten abundantly than argon they cannot strictly tial of xenon is actually lower than those of be called rare gases; this can be illustrated hydrogen, nitrogen, oxygen, fl uorine and by calculating the volumes occupied a.t s.t.p. -

Chemical Names and CAS Numbers Final
Chemical Abstract Chemical Formula Chemical Name Service (CAS) Number C3H8O 1‐propanol C4H7BrO2 2‐bromobutyric acid 80‐58‐0 GeH3COOH 2‐germaacetic acid C4H10 2‐methylpropane 75‐28‐5 C3H8O 2‐propanol 67‐63‐0 C6H10O3 4‐acetylbutyric acid 448671 C4H7BrO2 4‐bromobutyric acid 2623‐87‐2 CH3CHO acetaldehyde CH3CONH2 acetamide C8H9NO2 acetaminophen 103‐90‐2 − C2H3O2 acetate ion − CH3COO acetate ion C2H4O2 acetic acid 64‐19‐7 CH3COOH acetic acid (CH3)2CO acetone CH3COCl acetyl chloride C2H2 acetylene 74‐86‐2 HCCH acetylene C9H8O4 acetylsalicylic acid 50‐78‐2 H2C(CH)CN acrylonitrile C3H7NO2 Ala C3H7NO2 alanine 56‐41‐7 NaAlSi3O3 albite AlSb aluminium antimonide 25152‐52‐7 AlAs aluminium arsenide 22831‐42‐1 AlBO2 aluminium borate 61279‐70‐7 AlBO aluminium boron oxide 12041‐48‐4 AlBr3 aluminium bromide 7727‐15‐3 AlBr3•6H2O aluminium bromide hexahydrate 2149397 AlCl4Cs aluminium caesium tetrachloride 17992‐03‐9 AlCl3 aluminium chloride (anhydrous) 7446‐70‐0 AlCl3•6H2O aluminium chloride hexahydrate 7784‐13‐6 AlClO aluminium chloride oxide 13596‐11‐7 AlB2 aluminium diboride 12041‐50‐8 AlF2 aluminium difluoride 13569‐23‐8 AlF2O aluminium difluoride oxide 38344‐66‐0 AlB12 aluminium dodecaboride 12041‐54‐2 Al2F6 aluminium fluoride 17949‐86‐9 AlF3 aluminium fluoride 7784‐18‐1 Al(CHO2)3 aluminium formate 7360‐53‐4 1 of 75 Chemical Abstract Chemical Formula Chemical Name Service (CAS) Number Al(OH)3 aluminium hydroxide 21645‐51‐2 Al2I6 aluminium iodide 18898‐35‐6 AlI3 aluminium iodide 7784‐23‐8 AlBr aluminium monobromide 22359‐97‐3 AlCl aluminium monochloride -

3 • Molecules and Compounds
South Pasadena · AP Chemistry Name ____________________________________ Period ___ Date ___/___/___ 3 · Molecules and Compounds STUDY QUESTIONS and PROBLEMS 1. a. The structural formula for acetic acid is CH3CO2H. What is its empirical formula; what is its molecular formula? b. The molecular formula of acrylonitrile is C3H3N. Look up in the text, and draw, its structural formula. c. The molecular formula of aspartame (nutrasweet) is C14H18O5N2. Look up in the text, and draw, its structural formula. 2. The formulas for ethanol and ammonium nitrate are C2H5OH and NH4NO3. In what respects are these formulas and compounds different? 3. The molecular formula for both butanol and diethylether is C4H10O. Write structural formulas for both and show how they are different. Are any other structures possible? 4. Name the polyatomic ions: - - CH3CO2 HCO3 - 2- H2PO4 Cr2O7 2- - SO3 ClO 4 5. What are the formulas of the polyatomic ions: phosphate nitrite sulfate cyanide bisulfite chlorite 6. Write the ions present in the following salts and predict their formulas: potassium bromide calcium carbonate magnesium iodide lithium oxide aluminum sulfate ammonium chlorate beryllium phosphate 7. Name the following ionic salts: (NH4)2SO4 Co2(SO4)3 KHCO3 NiSO4 Ca(NO3)2 AlPO4 8. Name the following binary compounds of the nonmetals: CS2 SiCl4 SF6 GeH4 IF5 P4O10 N2H4 S4N4 PCl5 OF2 Cl2O7 IF7 9. What are the formulas for the following binary compounds? silicon dioxide phosphine boron trifluoride silicon carbide xenon tetroxide phosphorus tribromide dinitrogen pentoxide disulfur dichloride bromine trifluroide hydrogen selenide carbon tetrachloride 10. a. How many moles are present in 128 grams of sulfur dioxide? b. -

Xenon Iron Oxides Predicted As Potential Xe Hosts in Earth’S Lower Mantle
ARTICLE https://doi.org/10.1038/s41467-020-19107-y OPEN Xenon iron oxides predicted as potential Xe hosts in Earth’s lower mantle ✉ Feng Peng 1,2,8, Xianqi Song 3,4,8, Chang Liu4,5,6, Quan Li 3,4,5,6 , Maosheng Miao 2, ✉ ✉ Changfeng Chen 7 & Yanming Ma 3,4,5 An enduring geological mystery concerns the missing xenon problem, referring to the abnormally low concentration of xenon compared to other noble gases in Earth’s atmosphere. 1234567890():,; Identifying mantle minerals that can capture and stabilize xenon has been a great challenge in materials physics and xenon chemistry. Here, using an advanced crystal structure search algorithm in conjunction with first-principles calculations we find reactions of xenon with recently discovered iron peroxide FeO2, forming robust xenon-iron oxides Xe2FeO2 and XeFe3O6 with significant Xe-O bonding in a wide range of pressure-temperature conditions corresponding to vast regions in Earth’s lower mantle. Calculated mass density and sound velocities validate Xe-Fe oxides as viable lower-mantle constituents. Meanwhile, Fe oxides do not react with Kr, Ar and Ne. It means that if Xe exists in the lower mantle at the same pressures as FeO2, xenon-iron oxides are predicted as potential Xe hosts in Earth’s lower mantle and could provide the repository for the atmosphere’s missing Xe. These findings establish robust materials basis, formation mechanism, and geological viability of these Xe-Fe oxides, which advance fundamental knowledge for understanding xenon chemistry and physics mechanisms for the possible deep-Earth Xe reservoir. 1 College of Physics and Electronic Information & Henan Key Laboratory of Electromagnetic Transformation and Detection, Luoyang Normal University, 471022 Luoyang, China. -

Properties of Acids
Properties of Acids Solutions of acids have a sour taste Don’t taste them in the lab !!! 1 They change the colors of many indicators Acids turn blue litmus to red Acids turn bromothymol blue from blue to yellow They react with metals to generate hydrogen gas, H2 1 Metal Activity Series More active Li, K, Ca, Na, Mg, Al, Mn, Zn, Fe, Co, Ni, Pb, H, Cu, Hg, Ag, Pt, Au Less active Active enough to displace Cannot displace hydrogen from an acid hydrogen from an acid 2 Properties of Acids They react with metal oxides forming the salt of the metal and water CaO + 2HCl → CaCl2 + H2O They react with metal hydroxides forming the salt of the metal and water Ca(OH)2 + 2HCl → CaCl2 + 2H2O 3 Oxides Compounds of oxygen and another element There are two ways to name oxides Based on the oxidation number of the element Li2O – lithium oxide BaO – barium oxide FeO – iron(II) oxide Fe2O3 – iron(III) oxide Based on the number of atoms of each element Li2O – dilithium oxide BaO – barium oxide FeO – iron oxide Fe2O3 – diiron trioxide 4 Example 1 Name the following compounds: BeO, Al2O3, Cu2O, OsO4, Cr2O3, CrO3 5 Example 2 Write formulas for the following compounds: Potassium oxide Boron oxide Diindium trioxide Cobalt(II) oxide Dinitrogen pentoxide Rhenium(VI) oxide Xenon tetroxide Carbon monoxide Carbon dioxide Manganese(VII) oxide 6 Example 3 Write total and net ionic equations for the reaction between cobalt (III) oxide and diluted hydroiodic acid 7 Example 4 Write total and net ionic equations for the reaction between dialuminum -

Coordination Chemistry of Xenon
Coordination Chemistry of Xenon Paul Griffin Literature Seminar 11/14/19 Situated on the far side of the periodic table, the chemical inertness of the noble gases has led to their widespread applications ranging from gas-discharge lamps and ion engines for space travel to medical applications including as MRI contrast agents and radiotherapy.1 Though all practical applications of the noble gases center around Xe (0), understanding the reactivity and bonding of xenon in higher oxidation states is fundamental to investigating chemistry at the far end of the periodic table. The reactivity of the noble gases, in particular xenon, was first uncovered when Bartlett, who had previously prepared dioxygenyl hexafluoroplatinate (V) (O2PtF6), noted similar ionization energies for molecular oxygen and atomic xenon (a difference of 0.1 eV).2 Mixing of xenon with PtF6 vapor led to the formation of an orange-yellow solid led to the synthesis of the first noble gas compound, xenon hexafluoroplatinate (V) (XePtF6). The chemistry of xenon has evolved significantly since its’ discovery to include an assortment of xenon fluorides, oxides, oxylfluorides, organoxenon3, and metalloxenon compounds such as the tetraxenoaurate (II) cation (Figure 1).4 These species have weak covalent bonding between Xe and the oxygen/fluorine moieties.5 The chemistry of xenon is dominated by the 2+, 4+, and 6+ oxidation states, though Xe (VIII) compounds are known in the form of perxenates and xenon tetroxide. These primarily ionic species, particularly the oxides, oxylfluorides, and fluorides, serve as precursors to noncovalent adducts of xenon. Figure 1. Organoxenon and metalloxenon species Recent work has led to the preparation of xenon adducts with noncovalently interacting ligands such as acetonitrile6a, 6b, ketones6c, sulfoxides6c, and crown ethers6d (Figure 2) capable of weakly coordinating to xenon salts to afford stable species at room temperature. -

Chapter 20 the Representative Elements
Chapter 20 The Representative Elements Copyright ©2017 Cengage Learning. All Rights Reserved. Chapter 19 Table of Contents . (20.1) A survey of the representative elements . (20.2) The group 1A elements . (20.3) The chemistry of hydrogen . (20.4) The group 2A elements . (20.5) The group 3A elements . (20.6) The group 4A elements . (20.7) The group 5A elements . (20.8) The chemistry of nitrogen Copyright ©2017 Cengage Learning. All Rights Reserved. Chapter 19 Table of Contents . (20.9) The chemistry of phosphorus . (20.10) The group 6A elements . (20.11) The chemistry of oxygen . (20.12) The chemistry of sulfur . (20.13) The group 7A elements . (20.14) The group 8A elements Copyright ©2017 Cengage Learning. All Rights Reserved. Section 20.1 A Survey of the Representative Elements Reviewing the Periodic Table . Representative elements: Chemical properties are determined by the valence-level s and p electrons . Designated Groups 1A–8A . Transition metals: Result from the filling of d orbitals . Present at the center of the periodic table Copyright ©2017 Cengage Learning. All Rights Reserved. Copyright © Cengage Learning. All rights reserved 4 Section 20.1 A Survey of the Representative Elements Reviewing the Periodic Table (Continued) . Lanthanides: Correspond to the filling of the 4f orbitals . Actinides: Correspond to the filling of the 5f orbitals . Metalloids (semimetals): Elements along the division line between metals and nonmetals in the periodic table . Exhibit both metallic and nonmetallic characteristics Copyright ©2017 Cengage Learning. All Rights Reserved. Copyright © Cengage Learning. All rights reserved 5 Section 20.1 A Survey of the Representative Elements Differentiating Metals from Nonmetals Metals Nonmetals • Form cations by losing • Form anions by gaining valence electrons electrons • Exhibit electron • Exhibit electron configuration of the configuration of the noble gas from the noble gas in the same preceding period period Copyright ©2017 Cengage Learning. -

اختبار Naming Compounds / الأستاذ سعيد الشريف 0508168408
اختبار Naming compounds / اﻷستاذ سعيد الرشيف 0508168408 Ionic compounds # Question Your answer Result 1. tin (II) phosphite Sn3(PO3)2 2. mercury (I) chloride HgCl 3. cobalt (III) chlorite Co(ClO2)3 4. manganese (III) dichromate Mn2(Cr2O7)3 5. manganese (III) chlorite Mn(ClO2)3 6. barium sulfate BaSO4 7. KNO2 potassium nitrite 8. zinc nitride Zn3N2 9. CaCr2O7 calcium dichromate 10. silver sulfite Ag2SO3 11. CoC2O4 cobalt (II) oxalate 12. K4SiO4 potassium silicate 13. mercury (II) oxide HgO 14. cobalt (III) chromate Co2(CrO4)3 15. lead (II) oxalate PbC2O4 16. Sn(MnO4)4 tin (IV) permanganate 17. chromium (II) hydrogen sulfate Cr(HSO4)2 18. Sn(HCO3)4 tin (IV) hydrogen carbonate 19. cobalt (II) permanganate Co(MnO4)2 20. AgNO3 silver nitrate 21. FeP iron (III) phosphide 22. KHCO3 potassium hydrogen carbonate 23. strontium cyanide Sr(CN)2 24. lead (II) dichromate PbCr2O7 25. potassium oxide K2O 26. FeC2O4 iron (II) oxalate 27. aluminum acetate Al(C2H3O2)3 28. Co2(SO4)3 cobalt (III) sulfate مع حتيات/ اﻷستاذ سعيد الرشيف 0508168408 1 اختبار Naming compounds / اﻷستاذ سعيد الرشيف 0508168408 29. chromium (III) iodide CrI3 30. FeCl2 iron (II) chloride 31. mercury (I) hydrogen carbonate HgHCO3 32. Hg3(PO4)2 mercury (II) phosphate 33. Sn(HSO4)2 tin (II) hydrogen sulfate 34. Sn(MnO4)2 tin (II) permanganate 35. Al4C3 aluminum carbide 36. Sn(H2PO4)4 tin (IV) dihydrogen phosphate 37. Hg(OH)2 mercury (II) hydroxide 38. Al(H2PO4)3 aluminum dihydrogen phosphate 39. lead (IV) monohydrogen phosphate Pb(HPO4)2 40. Cr3N2 chromium (II) nitride 41. -
Chemistry Molecule Exercises
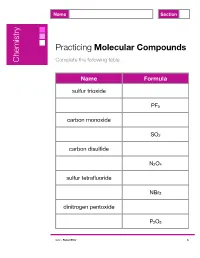
Name Section Practicing Molecular Compounds Chemistry Complete the following table. Name Formula sulfur trioxide PF5 carbon monoxide SO2 carbon disulfide N2O4 sulfur tetrafluoride NBr3 dinitrogen pentoxide P2O5 ©2017 Robert Prior A Name Answer Key Section Practicing Molecular Compounds Chemistry Complete the following table. Name Formula sulfur trioxide SO3 phosphorus pentafluoride PF5 carbon monoxide CO sulfur dioxide SO2 carbon disulfide CS2 dinitrogen tetroxide N2O4 sulfur tetrafluoride SF4 nitrogen tribromide NBr3 dinitrogen pentoxide N2O5 diphosphorus pentoxide P2O5 ©2017 Robert Prior A Name Section Practicing Molecular Compounds Chemistry Complete the following table. Name Formula ammonia SiO2 boron nitride CI4 molecular oxygen BCl3 ozone N2 diphosphorus pentasulfide H2O ©2017 Robert Prior B Name Answer Key Section Practicing Molecular Compounds Chemistry Complete the following table. Name Formula ammonia NH3 silicon dioxide SiO2 boron nitride BN carbon tetraiodide CI4 molecular oxygen O2 boron trichloride BCl3 ozone O3 molecular nitrogen N2 diphosphorus pentasulfide P2S5 water H2O ©2017 Robert Prior B Name Section Practicing Molecular Compounds Chemistry Complete the following table. Name Formula iodine Cl2 phosphorus tribromide ClF3 carbon disulfide N2O oxygen difluoride CO chlorine dioxide CF4 ©2017 Robert Prior C Name Answer Key Section Practicing Molecular Compounds Chemistry Complete the following table. Name Formula iodine I2 chlorine Cl2 phosphorus tribromide PBr3 chlorine trifluoride ClF3 carbon disulfide CS2 dinitrogen monoxide N2O oxygen difluoride OF2 carbon monoxide CO chlorine dioxide ClO2 carbon tetrafluoride CF4 ©2017 Robert Prior C Name Section Practicing Molecular Compounds Chemistry Complete the following table. Name Formula bromine CO disulfur decafluoride OF2 silicon monoxide PCl3 nitrogen dioxide SF6 xenon tetroxide SiCl4 ©2017 Robert Prior D Name Answer Key Section Practicing Molecular Compounds Chemistry Complete the following table. -

The Chemistry of Xenon
THE CHEMISTRY OF XENON' JOHN G. MALM, HENRY SELIG, Chemistry Division, Argonne National Laboratory, Argonne, Illinois JOSHUA JORTNER, AND STUART A. RICE Department of Chemistry and Institute for the Study of Metals, University of Chicago, Chicago, Illinois Received September 9, 1964 CONTENTS I. Introduction.. , . 200 11. The Xenon Fluorides. , , . 200 200 * . 201 203 206 4. XeFe.......................................,... ................. 206 5. XeOFI... .. , , , . , . 208 111. Complexes of Xenon.. ........................... 209 A. Xe(PtF&. ..... ..... .. ... ...... .. ....... 209 210 C. Xenon Difluoride Complexes, . 210 210 210 ............... 210 A. XeF ......................................................................... 210 .............. 210 210 D. XeOFs.. ............ ............................. 211 211 211 211 A. General.. .. .. ... ..... .. .. .. ... ... .. .............. .... ... 211 211 211 D. Xenon Trioxide ............................................................... 212 E. Properties of Aqueous Xe(VII1). ... 212 F. Oxidation Potentials. 213 G. Perxenate Salts.. , , . , . , . 213 H. Xenon Tetroxide.. .................... ........... ............. 213 I. Analysis for Xe(V1) and Xe(V1 Oxidation States., . 213 VI. The Nature of the Chemical Bond in the Xenon Fluorides.. , . , . , . 214 A. The Electron-Correlation Model, . , . , , . , , . , , . 214 B. The Hybridization Model.. ............ ............................. 216 C. Long-Range Xenon-Fluorine Interactions . , . , . , . , . 216 D. The Molecular Orbital Model. -
J. Rohonczy: Inorganic Chemistry I
Dr. János Rohonczy Lecture Notes Eötvös Loránd University, Budapest Faculty of Sciences Dr. János Rohonczy INORGANIC CHEMISTRY I. Lecture Notes Eötvös Loránd University Faculty of Sciences BUDAPEST 2017. János Rohonczy: Inorganic Chemistry I. Lecture Notes. Copyright © 2017 Dr. János Rohonczy, Eötvös Loránd University, Budapest, Faculty of Sciences All Right are Reserved. No part of this publication may be reproduced, stored in a retrieval system or transmitted in any form or by any means: electronic, electrostatic, magnetic tape, mechanical, photographical, photocopying, recording or otherwise, without permission in writing form the publisher. This book is written utilized the lecture notes of the Inorganic Chemistry lectures of the author at the Department of Inorganic Chemistry of Eötvös Loránd University, Budapest. Revised, and the fullerene and boron cluster topics remarked by Dr. Béla Csákvári professor emeritus. First edition 2017 Edited and cover page made by Dr. János Rohonczy Publisher: Eötvös Loránd University, Faculty of Sciences ISBN: 978-963-284-853-2 DOI: 10.21862/ 3 Table of Contents Introduction 7 1. Hydrogen 8 1.1. Hydrogen compounds 9 2. Halogens: F, Cl, Br, I, At 10 2.1. Hydrogen halides 13 2.2. Interhalogens 14 2.3. Polyhalogen and interhalogen ions, organic derivatives 16 3. (16th column) O, S, Se, Te, Po 17 3.1. Oxygen (O) 17 3.1.1. Oxygen compounds 19 3.1.2. Halogen oxides and oxygen halides 21 3.1.3. Halogen oxoacids and their salts 24 3.1.4. Halogen oxofluorides and fluorinated oxoacids 28 3.2. Sulfur(S) 29 3.2.1. Sulfur containing compounds 31 3.2.2.