Chromosome 15 in Floppy Infants Copyright
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Hypotonia and Lethargy in a Two-Day-Old Male Infant Adrienne H
Hypotonia and Lethargy in a Two-Day-Old Male Infant Adrienne H. Long, MD, PhD,a,b Jennifer G. Fiore, MD,a,b Riaz Gillani, MD,a,b Laurie M. Douglass, MD,c Alan M. Fujii, MD,d Jodi D. Hoffman, MDe A 2-day old term male infant was found to be hypotonic and minimally abstract reactive during routine nursing care in the newborn nursery. At 40 hours of life, he was hypoglycemic and had intermittent desaturations to 70%. His mother had an unremarkable pregnancy and spontaneous vaginal delivery. The mother’s prenatal serology results were negative for infectious risk factors. Apgar scores were 9 at 1 and 5 minutes of life. On day 1 of life, he fed, stooled, and voided well. Our expert panel discusses the differential diagnosis of hypotonia in a neonate, offers diagnostic and management recommendations, and discusses the final diagnosis. DRS LONG, FIORE, AND GILLANI, birth weight was 3.4 kg (56th PEDIATRIC RESIDENTS percentile), length was 52 cm (87th aDepartment of Medicine, Boston Children’s Hospital, d e percentile), and head circumference Boston, Massachusetts; and Neonatology Section, Medical A 2-day old male infant born at Genetics Section, cDivision of Child Neurology, and 38 weeks and 4 days was found to be was 33 cm (12th percentile). His bDepartment of Pediatrics, Boston Medical Center, Boston, limp and minimally reactive during physical examination at birth was Massachusetts routine care in the newborn nursery. normal for gestational age, with Drs Long, Fiore, and Gillani conceptualized, drafted, Just 5 hours before, he had an appropriate neurologic, cardiac, and and edited the manuscript; Drs Douglass, Fujii, and appropriate neurologic status when respiratory components. -

Hypotonia Surestep Product Catalog Page 29 in Step with Pediatric Hypotonia
SPECIAL EDUCATIONAL SERIES DIAGNOSTIC INSIGHTS ANALYZING GAIT CHANGES GROSS MOTOR SKILLS ORTHOTIC MANAGEMENT CLI N I CAL CASE STUDIES Sponsored by an educational grant from: In Step With Pediatric Hypotonia SureStep Product Catalog Page 29 In Step With Pediatric Hypotonia Contents VIEWPOINT FROM THE EDITOR: An Unexpected Path, Mobility and More an Invaluable Perspective At the most basic level, mobility is about get- PAGE 3 ting from point A to point B. But, for many children with hypotonia, it’s about so much 4 more. FEATURES It’s about independence. It’s about con- fidence. It’s about maintaining strength, fit- ness, and healthy bones. It’s about not being Understanding Hypotonia excluded from activities enjoyed by their PAGE 4 typically developing peers. And improved mobility may have even Gait: The Cornerstone more benefits in those children whose hy- potonia is associated with social and behav- of Intervention ioral developmental delays. New research PAGE 8 has identified an association between motor skills and sociobehavioral milestones in chil- 8 The Importance of Gross dren with autism spectrum disorder, who often present with hypotonia (see “The Im- Motor Skills portance of Gross Motor Skills,” page 12). PAGE 12 This suggests that early intervention to improve gross motor skills—including or- thotic devices and physical therapy—may Orthotic Solutions for also help certain children interact more Children with Hypotonia comfortably with others. That won’t come as PAGE 16 a surprise to the clinicians and parents who 12 have personally seen it happen. This special issue is filled with evidence- Orthotic Success Stories: based information and personal success sto- Four Cases in a Series ries illustrating how effective interventions can enhance mobility in children with hy- PAGE 20 potonia. -

Impact of Fluorescence in Situ Hybridization in the Detection of Cryptic Fusion Transcript PML/RARA and a Complex T(5;15;17) in a Case of Acute Promyelocytic Leukemia
Impact of Fluorescence in Situ Hybridization in the Detection of Cryptic Fusion Transcript PML/RARA and A Complex t(5;15;17) in a Case of Acute Promyelocytic Leukemia Gönül OGUR*,****, Turgay FEN**, Gülsan SUCAK***, Pierre HEIMANN*, Gaye CANKUÞ****, Rauf HAZNEDAR**** * Univérsité Libre de Bruxelles, Hôpital Erasme, Service Génétique Medicale, Bruxelles, BELGIUM ** Oncology Hospital, Ankara, TURKEY *** Adult Hematology Department, Faculty of Medicine, Gazi University, Ankara, TURKEY **** GEN-MED Genetic Diseases and Prenatal Diagnosis Center, Ankara, TURKEY ABSTRACT Genetic aspects of a 28 year-old female patient with typical morphological and clinical features of acute promyelocytic leukemia is presented. Pml/rara fusion transcript and a complex translocation involving chromosomes 5, 15 and 17 were detected by fluorescence in situ hybridization (FISH) tech- nique which was applied as in adjunct to conventional cytogenetics. The patient deceased soon in spite of the immediate ATRA and cytostatic therapy. Key Words: FISH, PML/RARA, Acute promyelocytic leukemia. Turk J Haematol 2000;17(4):207-212. INTRODUCTION sociated with t(15;17)(q22;q12-21). The diagnosis of APL and the detection of residual disease are Acute promyelocytic leukemia (APL) is a rara based on the presence of this translocati- distinct subtype of myeloid leukemia characterized on[1,3,4,5,6,7,15]. Rarely alternative balanced trans- by invasion of bone marrow by hypergranular le- locations have been described in subtypes of APL, ukemia cells, by specific translocations almost al- and as more cases are being evaluated, complex, ways involving chromosome 17 and by a high sen- variant translocations are increasingly recogni- sitivity of the promyelocytic blasts to retinoic acid zed[17,19,20,22, 24]. -
IUGR: Intrauterine Growth Restriction
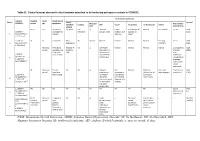
Table S1. Clinical features observed in the 6 patients described so far harboring pathogenic variants in FOXRED1. Evolutionary symptoms Variants Prenatal Onset Onset clinical Patient Lactic/ Survival FOXRED1 period age symptoms Muscular Psychomotor Metabolic Epilepsy MRI Visual Respiratory Cardiovascular Others tone development acidosis IUGR 2m Hypotonia Yes (+++) Yes ↓ Normal Latent Bronchospasm Normal AEP normal IQ: 48 Alive c.920G>A Development refractary (2m,4y,7y3m) strabismus of episodes in (15y) 1 (p.Gly307Glu) / al delay right eye infant c.733+1G>A c.920G>A NI 4y Clumsiness With No Normal Normal Normal Normal Normal Learning IQ: 99 Alive 2 (p.Gly307Glu) / exercise (+) disorders (19y) c.733+1G>A - Neonatal Premature; No (only ↑ Yes ↓ Decreased Normal Normal Normal Normal Gradually loss Alive period Hypoglycemia lactate in attenuation in of motor (22y)) Congenital LCR) the putamen skills; c.694C>T lactic acidosis and cerebellar wheelchair; (p.Gln232X) / 3 atrophy (6y) no expressive c.1289A>G language; 18 (p.Asn430Ser) understands simple commands NI Neonatal Truncal Yes Yes ↓ Delayed Eye Normal Mild non- Persistent Psychomotor Alive period hypotonia myelination movements obstructive left hepatomegaly retardation (10y) c.1054 C>T Poor feeding ventricular have always ventricular (p.Arg352Trp) / dilatation; been roving hypertrophy 4 c.1054 C>T abnormal signal bilateral optic (p.Arg352Trp) 19 in the thalami atrophy and basal ganglia (8m) c.1308G>A ND ND ND ND Yes NA NA NA NA NA NA Severe Alive (p.Val421Met) / psychomotor (¿) 5 c.1308G>A retardation (p.Val421Met)20 IUGR; Neonatal Congenital Yes Yes ↓ Large -- Persistent Dilated right - - Death (3 c.612_615dupA Cerebral period lactic periventricular severe ventricle and months) CTG intraventric acidosis. -

Cerebral Hypotonia by Mihee Bay MD (Dr
Cerebral hypotonia By Mihee Bay MD (Dr. Bay of Kennedy Krieger Institute and Johns Hopkins School of Medicine has no relevant financial relationships to disclose.) Originally released July 12, 2006; last updated February 1, 2016; expires February 1, 2019 Introduction This article includes discussion of cerebral hypotonia, central hypotonia, essential hypotonia, benign congenital hypotonia, and floppy infant. The foregoing terms may include synonyms, similar disorders, variations in usage, and abbreviations. Overview Hypotonia is a clinical manifestation of numerous diseases affecting the central and/or peripheral motor nervous system. The key to accurate diagnosis involves integral steps of evaluation that include a detailed history, examination, and diagnostic tests. “Cerebral” (or central) hypotonia implies pathogenesis from abnormalities from the central nervous system, and related causal disorders include cerebral dysgenesis and genetic or metabolic disorders. Patients with central hypotonia generally have hypotonia without associated weakness, in contrast to the peripheral (lower motor neuron) causes, which typically produce both hypotonia and muscle weakness. Hypotonia is a clinical manifestation of over 500 genetic disorders; thus, a logical, stepwise approach to diagnosis is essential. With recent advances in the field of genetic testing, diagnostic yield will undoubtedly improve. There is no cure, but treatment includes supportive therapies, such as physical and occupational therapy, and diagnosis-specific management. Key points • Hypotonia is reduced tension or resistance of passive range of motion. • The first step in the evaluation of a child with hypotonia is localization to the central (“cerebral”) or peripheral nervous system, or both. • Central hypotonia is more likely to be noted axially with normal strength and hyperactive to normal deep tendon reflexes. -

Mean Platelet Volume in Asymptomatic Chorioamnionitis-Exposed Infants
www.jpnim.com Open Access eISSN: 2281-0692 Journal of Pediatric and Neonatal Individualized Medicine 2021;10(1):e100132 doi: 10.7363/100132 Received: 2019 Aug 22; revised: 2020 Jan 26; accepted: 2020 Feb 02; published online: 2020 Dec 28 Original article Mean platelet volume in asymptomatic chorioamnionitis- exposed infants. A retrospective case-control study Atef Alshafei, Moustafa Hassan, Yaser El saba, Anwar Khan, Mahmoud Ahmed Neonatology Section, Pediatric Department, Dubai Hospital, Dubai, UAE Abstract Introduction: Maternal chorioamnionitis (CA) is a serious condition causing several neonatal morbidities and long-term neurodevelopmental sequelae in exposed infants. Current guidelines still recommend admission, laboratory evaluation, and antibiotic administration to all CA-exposed infants. The incidence of early-onset neonatal sepsis (EOS) is currently low, owing to the routine intrapartum antibiotic administration to mothers identified to be at risk of developing CA. New diagnostic tools for early diagnosis of sepsis in apparently healthy infants exposed to maternal CA are needed. Previous studies showed that mean platelet volume (MPV) is evolving as a potential inflammatory marker of neonatal sepsis. We aimed to study whether MPV can be used as an adjuvant diagnostic tool for EOS in asymptomatic CA-exposed infants. Objective: To evaluate the role of MPV as an adjuvant biomarker of EOS in cases of asymptomatic CA-exposed infants. Design: Retrospective case-control study. Setting: A tertiary care Neonatal Intensive Care Unit (NICU). Patients: Asymptomatic CA-exposed infants 37-40 weeks of gestation admitted between May 2016 and April 2019 to the NICU of Dubai Hospital, UAE. Results: A total of 1,300 infants were admitted to NICU during the study period. -

Long-Term Outcome After Neonatal Meconium Obstruction
Long-term Outcome After Neonatal Meconium Obstruction Julie R. Fuchs, MD, and Jacob C. Langer, MD ABSTRACT. Objective. It is unclear whether children meconium ileus and those undergoing resection or enter- with cystic fibrosis (CF) who present with neonatal ostomy. Patients with meconium obstruction who do not meconium ileus have a different long-term outcome from have CF have an excellent long-term prognosis. This those presenting later in childhood with pulmonary com- information will be useful in counseling the families of plications or failure to thrive. We examined a cohort of infants presenting with neonatal meconium obstruction. patients with meconium ileus, and compared their long- Pediatrics 1998;101(4). URL: http://www.pediatrics.org/ term outcome with children who had CF without meco- cgi/content/full/101/4/e7; cystic fibrosis, meconium ileus, nium ileus and neonates who had meconium obstruction meconium plug syndrome. without CF (meconium plug syndrome). Study Design. Comparative study using retrospective and follow-up interview data. ABBREVIATION. CF, cystic fibrosis. Patients. Group 1 consisted of 35 surviving CF pa- tients who presented with meconium ileus between 1966 econium obstruction in the neonate is a and 1992. Two control groups were also studied: 35 age- spectrum of disease that includes meco- and sex-matched CF patients without meconium ileus 1 (group 2), and 12 infants presenting with meconium plug Mnium ileus and meconium plug syndrome. syndrome during the same time period (group 3). Meconium ileus is characterized by extremely viscid, Outcome Measures. Pulmonary, gastrointestinal, nu- protein-rich inspissated meconium causing terminal tritional, and functional status were reviewed, and sur- ileal obstruction, and accounts for approximately gical complications were recorded. -

Rapid Molecular Assays to Study Human Centromere Genomics
Downloaded from genome.cshlp.org on September 26, 2021 - Published by Cold Spring Harbor Laboratory Press Method Rapid molecular assays to study human centromere genomics Rafael Contreras-Galindo,1 Sabrina Fischer,1,2 Anjan K. Saha,1,3,4 John D. Lundy,1 Patrick W. Cervantes,1 Mohamad Mourad,1 Claire Wang,1 Brian Qian,1 Manhong Dai,5 Fan Meng,5,6 Arul Chinnaiyan,7,8 Gilbert S. Omenn,1,9,10 Mark H. Kaplan,1 and David M. Markovitz1,4,11,12 1Department of Internal Medicine, University of Michigan, Ann Arbor, Michigan 48109, USA; 2Laboratory of Molecular Virology, Centro de Investigaciones Nucleares, Facultad de Ciencias, Universidad de la República, Montevideo, Uruguay 11400; 3Medical Scientist Training Program, University of Michigan, Ann Arbor, Michigan 48109, USA; 4Program in Cancer Biology, University of Michigan, Ann Arbor, Michigan 48109, USA; 5Molecular and Behavioral Neuroscience Institute, University of Michigan, Ann Arbor, Michigan 48109, USA; 6Department of Psychiatry, University of Michigan, Ann Arbor, Michigan 48109, USA; 7Michigan Center for Translational Pathology and Comprehensive Cancer Center, University of Michigan Medical School, Ann Arbor, Michigan 48109, USA; 8Howard Hughes Medical Institute, Chevy Chase, Maryland 20815, USA; 9Department of Human Genetics, 10Departments of Computational Medicine and Bioinformatics, University of Michigan, Ann Arbor, Michigan 48109, USA; 11Program in Immunology, University of Michigan, Ann Arbor, Michigan 48109, USA; 12Program in Cellular and Molecular Biology, University of Michigan, Ann Arbor, Michigan 48109, USA The centromere is the structural unit responsible for the faithful segregation of chromosomes. Although regulation of cen- tromeric function by epigenetic factors has been well-studied, the contributions of the underlying DNA sequences have been much less well defined, and existing methodologies for studying centromere genomics in biology are laborious. -

Common Anomalies Associated To
ISSN: 2643-3885 Muhsin. Int J Foot Ankle 2018, 2:013 Volume 2 | Issue 2 Open Access International Journal of Foot and Ankle RESEARCH ARTICLE Common Anomalies Associated To Congenital Vertical Talus: A Single Center Experience Elmas Muhsin* Check for Department of Medical Genetic, Afyon Kocatepe University, Turkey updates *Corresponding author: Elmas Muhsin, Department of Medical Genetic, Afyon Kocatepe University, Afyonkarahisar, Turkey vicular. The incidence is estimated to be one in 10,000. Abstract Approximately half of all cases (idiopathic) are associ- Background: Congenital vertical talus is defined as a foot deformity in which the calcaneus is in equinus, the talus is ated with deformity and 2-5 neuromuscular and genet- plantarflexed, and there is a rigid and irreducible dislocation ic disorders in the remaining cases. There is evidence of the talonavicular joint complex, with the navicular articu- that some isolated deformities are transmitted as an lating on the dorsolateral aspect of the talar neck. It is often autosomal dominant feature with incomplete pene- associated with systemic involvement. trance [1-4]. The deformity is also known by congenital Aims: To identify the most common anomalies accompany- “rocker-bottom” flatfoot because of its rigid deformity ing to CVT (Congenital Vertical Talus). No literature investi- gating similar clinical data was found in the literature review. with the forefoot dorsiflexed and the hindfoot plantar- Study design: CVT has a systemic effect and is accom- flexed. The term “congenital convex pes valgus” is also panied by many anomalies. At the same time as this study, frequently used. To make a definite diagnosis, it is im- anomalies were frequently found accompanying CVT. -

Factors Associated with Low Muscle Tone and Impact of Common
Original Article DOI: 10.7860/JCDR/2019/39551.12675 Factors Associated with Low Muscle Tone and Section Physiotherapy Impact of Common Musculoskeletal Problems on Motor Development in Preterm Infants at One Year of Corrected Age SRIVARSHA TELEDEVARA1, M RAJESWARI2, R SIVA KUMAR3, N UDAYAKUMAR4 ABSTRACT motor development. Backward multiple regression, Chi-square Introduction: Structural immaturity of muscular system in test Pearson’s correlation were used for data analysis. Preterm Infants (PTI) results in maturation related hypotonia Results: Backward multiple regression analysis showed which is found to be influenced by risk factors present at the statistically significant association of Birth weight, Gestational time of birth. Low muscle tone can lead to lower extremity Age, and Apgar score with low muscle tone in PTI (p<0.05). Chi- malalignment, abnormal positioning and loading resulting in square test was used to compare the muscle tone of PTI and FTI musculoskeletal problems that could have an impact on motor which showed that ATA of PTI was significantly higher than FTI development which is not well established. but within physiological limits (p<0.05). Pearson’s correlation Aim: This study was carried out to analyse the risk factors coefficient showed that there is statistically significant positive associated with low muscle tone in PTI and the impact of correlation between muscle tone and musculoskeletal problems common musculoskeletal problems on motor development of and a negative correlation between musculoskeletal problems PTI at 1 year of corrected age. and Gross Motor Quotient (GMQ) of PDMS 2 in PTI at 1 year of corrected age (p<0.05). Materials and Methods: This Cross sectional study was carried out in 36 PTI and 36 Full Term Infants (FTI) who were recruited Conclusion: Maturation related hypotonia carried during the first from Child Development unit and the details of risk factors were year of life brings about musculoskeletal problems which have obtained from the records. -

WHO Recommendations on Interventions to Improve Preterm Birth Outcomes
WHO recommendations on interventions to improve preterm birth outcomes For more information, please contact: Department of Reproductive Health and Research World Health Organization ISBN 978 92 4 150898 8 Avenue Appia 20, CH-1211 Geneva 27, Switzerland Fax: +41 22 791 4171 E-mail: [email protected] www.who.int/reproductivehealth WHO recommendations on interventions to improve preterm birth outcomes WHO Library Cataloguing-in-Publication Data WHO recommendations on interventions to improve preterm birth outcomes. Contents: Appendix: WHO recommendations on interventions to improve preterm birth outcomes: evidence base 1.Premature Birth – prevention and control. 2.Infant, Premature. 3.Infant Mortality – prevention and control. 4.Prenatal Care. 5.Infant Care. 6.Guideline. I.World Health Organization. ISBN 978 92 4 150898 8 (NLM Classifi cation: WQ 330)Contents © World Health Organization 2015 All rights reserved. Publications of the World Health Organization are available on the WHO web site (www.who.int) or can be purchased from WHO Press, World Health Organization, 20 Avenue Appia, 1211 Geneva 27, Switzerland (tel.: +41 22 791 3264; fax: +41 22 791 4857; e-mail: [email protected]). Requests for permission to reproduce or translate WHO publications –whether for sale or for non-commercial distribution– should be addressed to WHO Press through the WHO website (www.who.int/about/licensing/copyright_form/en/index.html). The designations employed and the presentation of the material in this publication do not imply the expression of any opinion whatsoever on the part of the World Health Organization concerning the legal status of any country, territory, city or area or of its authorities, or concerning the delimitation of its frontiers or boundaries. -

Neonatal Hypotonia
Neonatal Hypotonia Clinical Approach to Floppy Baby Hypotonia in the newborn is a common presenting feature of systemic illness or neurologic dysfunction at any level of the central or peripheral nervous system. It is defined as reduced resistance to passive range of motion in joints. Etiology: diverse Causes include (but are not limited to): Central Sepsis (most common) Hypoxic ischemic encephalopathy Intracranial hemorrhage Cerebral malformations Chromosomal abnormalities (e.g.Trisomy 21, Prader-Willi syndrome) Congenital infections (TORCH) Drug effects (e.g. benzodiazepines, Magnesium toxicity) Inborn errors of metabolism Endocrine: hypothyroidism Benign congenital hypotonia Spinal cord Birth trauma (especially Breech delivery) Syringomyelia Anterior Horn Cell Spinal Muscular Atrophy Neurogenic arthrogryposis Neuromuscular Myasthenia gravis (transient/ congenital) junction Infantile botulism Peripheral nerves Congenital hypomyelinating neuropathy Hereditary motor and sensory neuropathies (Dejerine-Sottas disease) Hereditary sensory and autonomic neuropathy Guillain Barre syndrome (very rare) Muscle Congenital myopathies (e.g. central core disease, Nemaline Rod myopathy, myotubular myopathy, congenital fiber type disproportion and multicore myopathy) Congenital muscular dystrophies( merosin deficient, Walker- Warburg disease, muscle-eye-brain disease, Fukuyama disease Muscular dystrophies (inc. congenital myotonic dystrophy) Metabolic myopathies Disease of Glycogen Metabolism and multisystem Acid maltase deficiency